

Development of a Novel Antibacterial Peptide, PAM-5, via Combination of Phage Display Selection and Computer-Assisted Modification

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Phage Display Selection of Peptides Binding to P. aeruginosa
2.3. Phage-ELISA Screening for Phage Peptides Binding to P. aeruginosa
2.4. Phage Genomic DNA Sequencing and Peptide Determination
2.5. Peptide Modification and Synthesis
2.6. Antibacterial Effect of PAM-5 In Vitro
2.7. Antibacterial Effect of PAM-5 in Human Plasma
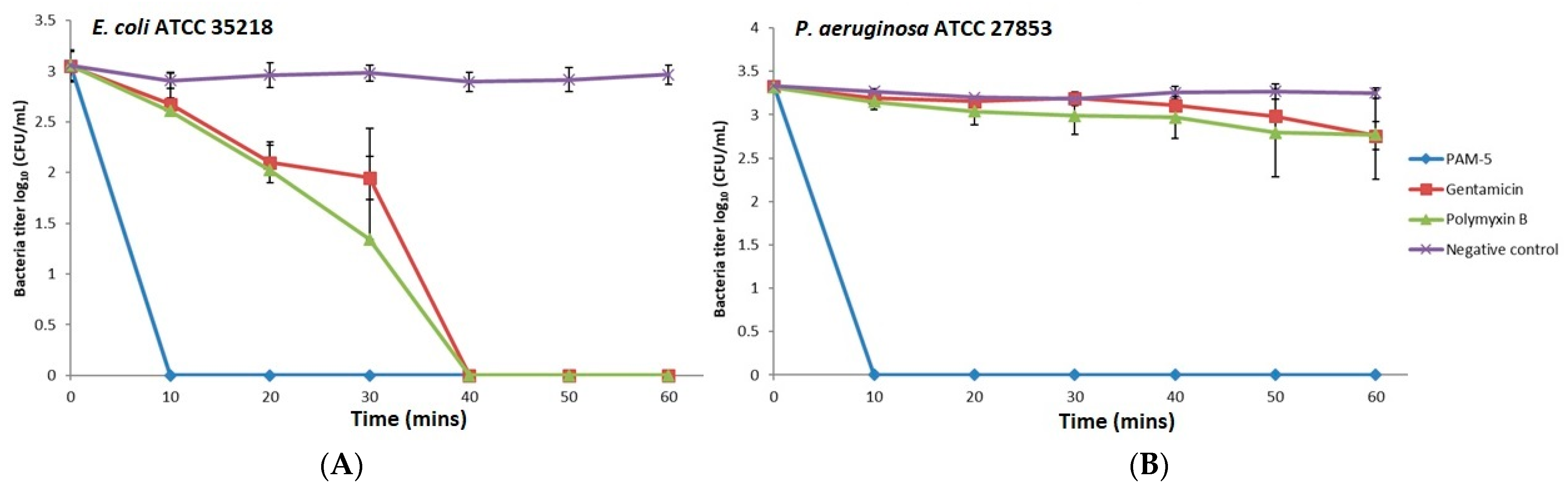
2.8. Time-Kill Assay
2.9. Scanning Electron Microscopy (SEM) on PAM-5-Treated Bacteria
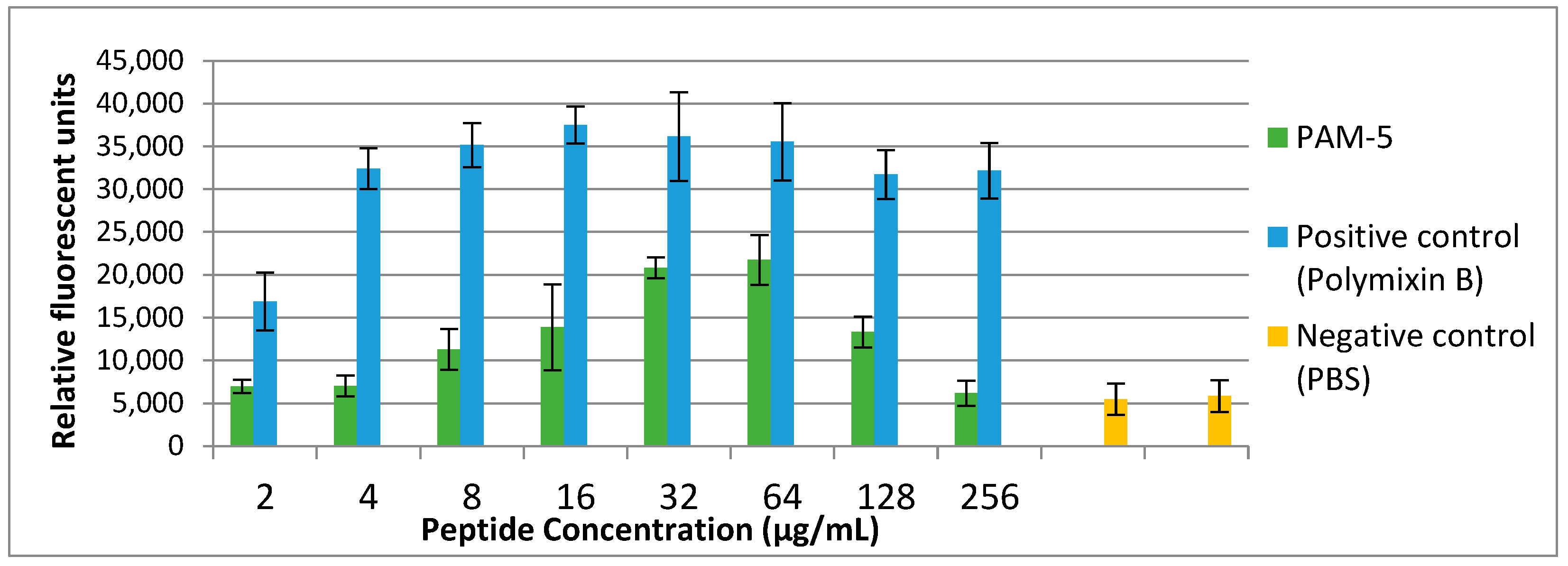
2.10. Membrane Permeabilization by PAM-5 via SYTOX Green Uptake Assay
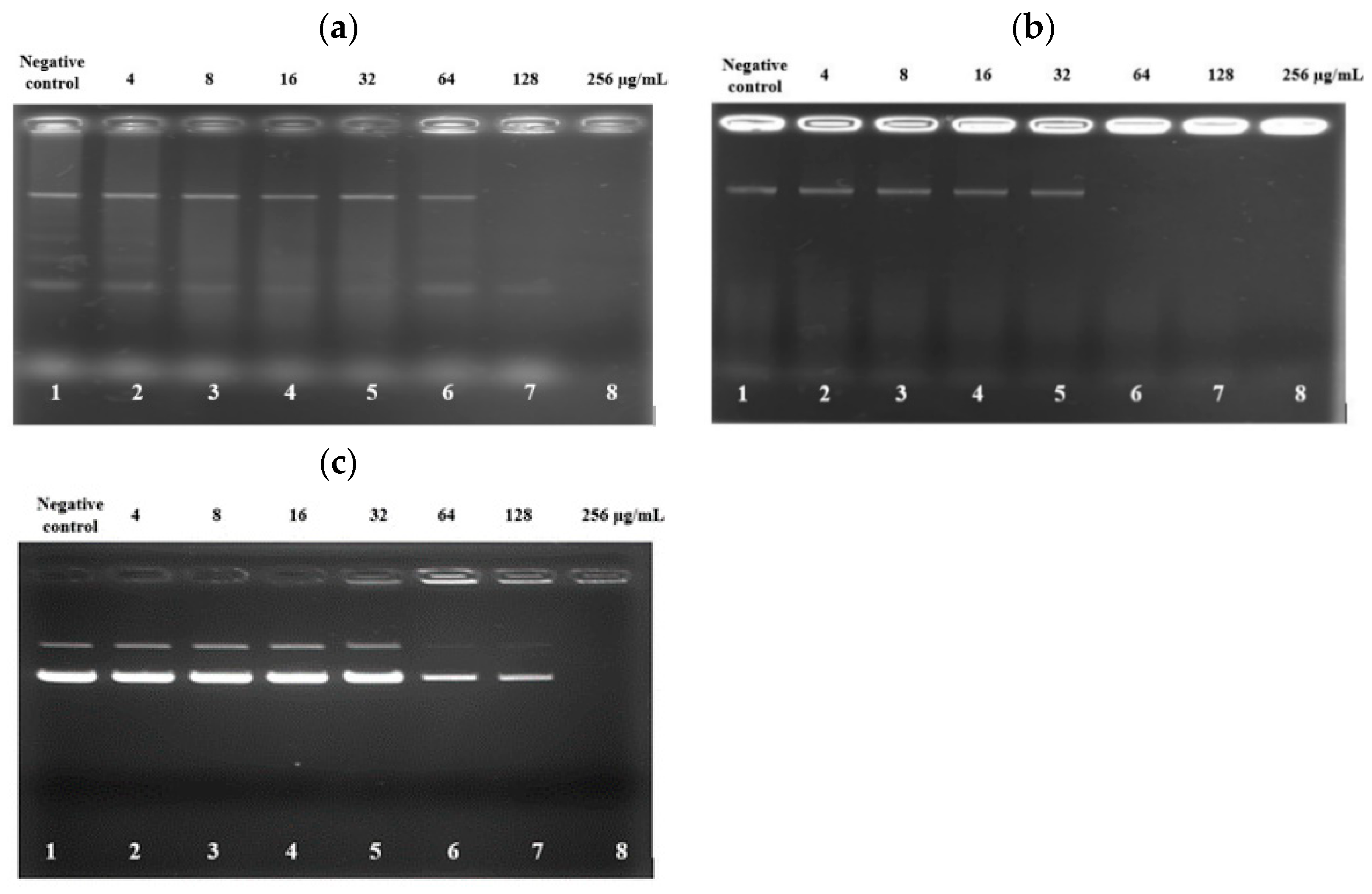
2.11. DNA Retardation Assay
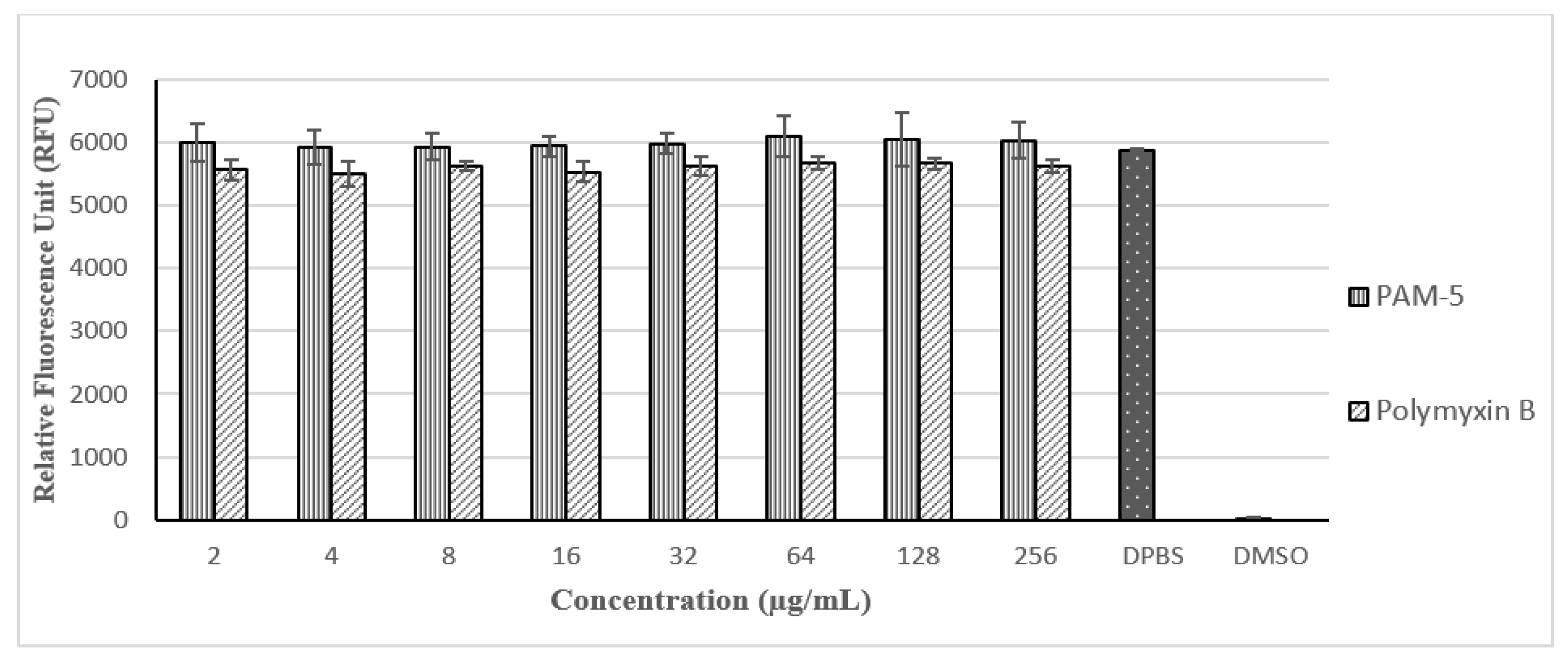
2.12. Cell Cytotoxicity Assay
2.13. Haemolytic Effect of PAM-5
2.14. Statistical Analysis
3. Results
3.1. Affinity-Selected Peptides Binding to P. aeruginosa
3.2. Peptide Modification
3.3. Antibacterial Effect of PAM-5
3.4. Stability of PAM-5 in Human Plasma
3.5. Killing Kinetics of PAM-5
3.6. SEM Examination on Morphology of PAM-5-Treated Bacteria
3.7. Cytoplasmic Membrane Permeabilization by PAM-5
3.8. PAM-5 Binding to Bacterial Genomic DNA and Plasmid DNA
3.9. Toxicity Effects of PAM-5 on Vero Cells
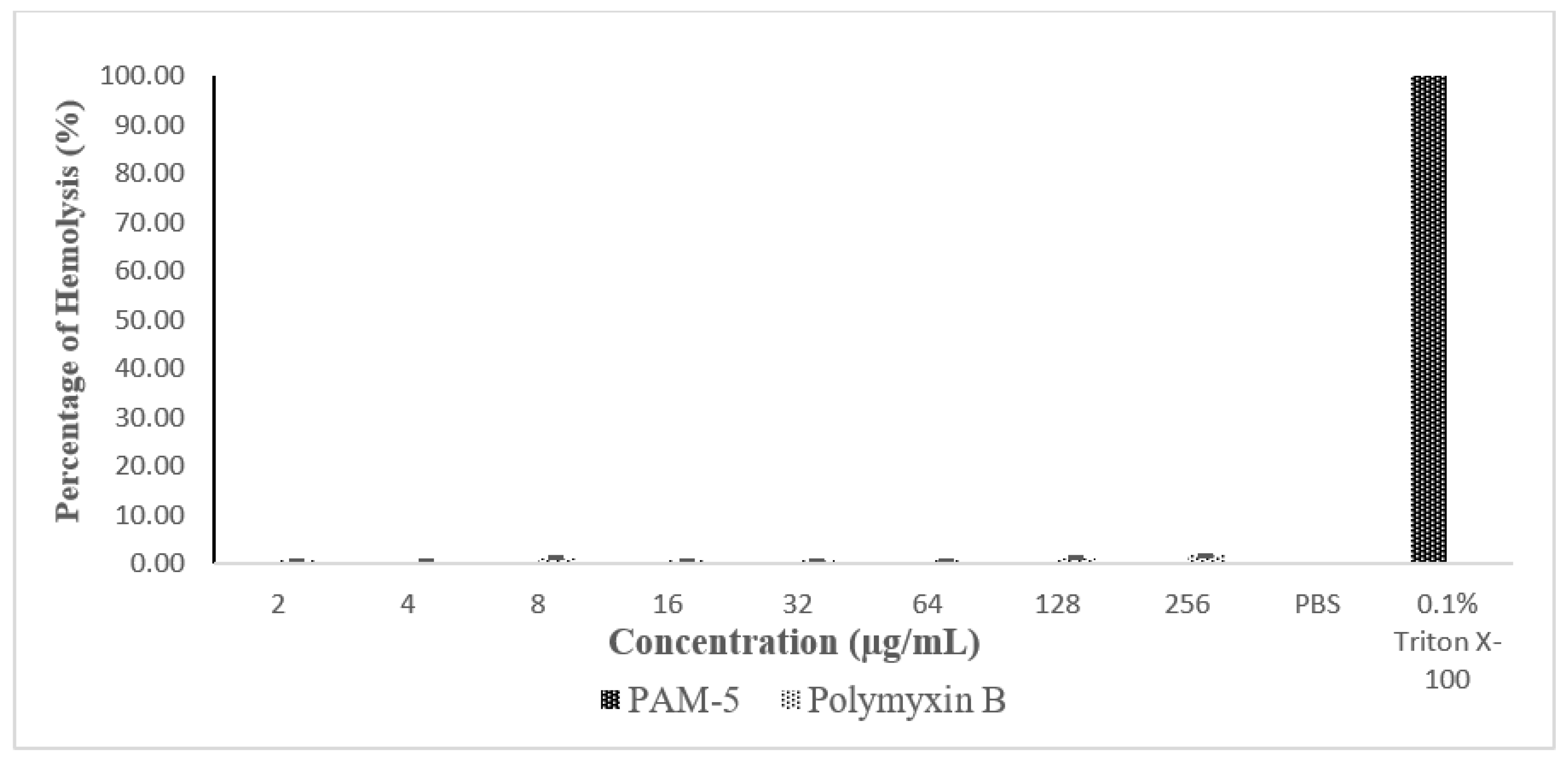
3.10. Haemolytic Effect of PAM-5 to hRBCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manohar, P.; Loh, B.; Nachimuthu, R.; Hua, X.; Welburn, S.C.; Leptihn, S. Secondary Bacterial Infections in Patients with Viral Pneumonia. Front. Med. 2020, 7, 420. [Google Scholar] [CrossRef] [PubMed]
- Feldman, C.; Anderson, R. The role of co-infections and secondary infections in patients with COVID-19. Pneumonia 2021, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.H.; Saha, B.K.; Ramani, A.; Chopra, A. State-of-the-art review of secondary pulmonary infections in patients with COVID-19 pneumonia. Infection 2021, 49, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Poirel, L. Epidemiology and Diagnostics of Carbapenem Resistance in Gram-negative Bacteria. Clin. Infect. Dis. 2019, 69 (Suppl. 7), S521–S528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brink, A.J. Epidemiology of carbapenem-resistant Gram-negative infections globally. Curr. Opin. Infect. Dis. 2019, 32, 609–616. [Google Scholar] [CrossRef]
- de Steenwinkel, J.E.; de Knegt, G.J.; ten Kate, M.T.; van Belkum, A.; Verbrugh, H.A.; Kremer, K.; van Soolingen, D.; Bakker-Woudenberg, I.A. Time-kill kinetics of anti-tuberculosis drugs, and emergence of resistance, in relation to metabolic activity of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2010, 65, 2582–2589. [Google Scholar] [CrossRef]
- Johnson, P.J.; Levin, B.R. Pharmacodynamics, population dynamics, and the evolution of persistence in Staphylococcus aureus. PLoS Genet. 2013, 9, e1003123. [Google Scholar] [CrossRef] [Green Version]
- Zahir, T.; Wilmaerts, D.; Franke, S.; Weytjens, B.; Camacho, R.; Marchal, K.; Hofkens, J.; Fauvart, M.; Michiels, J. Image-Based Dynamic Phenotyping Reveals Genetic Determinants of Filamentation-Mediated β-Lactam Tolerance. Front. Microbiol. 2020, 11, 374. [Google Scholar] [CrossRef] [Green Version]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.M.; Masuda, I.; Foster, L.J. tRNA methylation: An unexpected link to bacterial resistance and persistence to antibiotics and beyond. Wiley Interdiscip. Rev. RNA 2020, 11, e1609. [Google Scholar] [CrossRef] [PubMed]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrot, R.; Barylski, J.; Nowicki, G.; Broniarczyk, J.; Buchwald, W.; Goździcka-Józefiak, A. Plant antimicrobial peptides. Folia. Microbiol. 2014, 59, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Lei, M.; Du, X.; Cui, P.; Zhang, S. Identification of a novel antimicrobial peptide from amphioxus Branchiostoma japonicum by in silico and functional analyses. Sci. Rep. 2015, 5, 18355. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.H.; Jang, A.Y.; Lin, S.; Lim, S.; Kim, D.; Park, K.; Han, S.M.; Yeo, J.H.; Seo, H.S. Melittin, a honeybee venom derived antimicrobial peptide, may target methicillin resistant Staphylococcus aureus. Mol. Med. Rep. 2015, 12, 6483–6490. [Google Scholar] [CrossRef] [Green Version]
- de Barros, E.; Gonçalves, R.M.; Cardoso, M.H.; Santos, N.C.; Franco, O.L.; Cândido, E.S. Snake Venom Cathelicidins as Natural Antimicrobial Peptides. Front. Pharmacol. 2019, 10, 1415. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, A.; Machini, M.T. Antimicrobial lipopeptides: In vivo and in vitro synthesis. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Formatex Research Center: Badajoz, Spain, 2013; pp. 951–959. [Google Scholar]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.E.; Hancock, R.E.; Kalman, D. Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 2010, 31, 1957–1965. [Google Scholar] [CrossRef] [Green Version]
- Laverty, G.; Gilmore, B. Cationic antimicrobial peptide cytotoxicity. SOJ Microbiol. Infect. Dis. 2014, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Roberts, K.D.; Azad, M.A.; Wang, J.; Horne, A.S.; Thompson, P.E.; Nation, R.L.; Velkov, T.; Li, J. Antimicrobial Activity and Toxicity of the Major Lipopeptide Components of Polymyxin B and Colistin: Last-line Antibiotics against Multidrug-Resistant Gram-negative Bacteria. ACS Infect. Dis. 2015, 1, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Inui Kishi, R.N.; Stach-Machado, D.; Singulani, J.L.; Dos Santos, C.T.; Fusco-Almeida, A.M.; Cilli, E.M.; Freitas-Astúa, J.; Picchi, S.C.; Machado, M.A. Evaluation of cytotoxicity features of antimicrobial peptides with potential to control bacterial diseases of citrus. PLoS ONE 2018, 13, e0203451. [Google Scholar] [CrossRef]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The multifaceted nature of antimicrobial peptides: Current synthetic chemistry approaches and future directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.M.; Viñas, M. Future prospects for antimicrobial peptide development: Peptidomimetics and antimicrobial combinations. Expert. Opin. Drug Discov. 2021, 16, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Hurley, S.L.; Schmidt, F.J.; Erwin, A.L.; Smith, A.L. Peptides selected for binding to a virulent strain of Haemophilus influenzae by phage display are bactericidal. Antimicrob. Agents Chemother. 2005, 49, 2972–2978. [Google Scholar] [CrossRef] [Green Version]
- Bishop-Hurley, S.L.; Rea, P.J.; McSweeney, C.S. Phage-displayed peptides selected for binding to Campylobacter jejuni are antimicrobial. Protein Eng. Des. Sel. 2010, 10, 751–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pini, A.; Giuliani, A.; Falciani, C.; Runci, Y.; Ricci, C.; Lelli, B.; Malossi, M.; Neri, P.; Rossolini, G.M.; Bracci, L. Antimicrobial activity of novel dendrimeric peptides obtained by phage display selection and rational modification. Antimicrob. Agents Chemother. 2005, 49, 2665–2672. [Google Scholar] [CrossRef] [Green Version]
- Sainath Rao, S.; Mohan, K.V.; Atreya, C.D. A peptide derived from phage display library exhibits antibacterial activity against E. coli and Pseudomonas aeruginosa. PLoS ONE 2013, 8, e56081. [Google Scholar] [CrossRef] [Green Version]
- New England Biolabs. Applications of the Ph.D. Phage Display Peptide Libraries. 2020. Available online: https://international.neb.com/tools-and-resources/feature-articles/applications-of-the-phd-phage-display-peptide-libraries (accessed on 15 February 2020).
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 9th ed.; Approved Standard; CLSI document M07-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Cockerill, F. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Second Informational Supplement; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012; ISBN 978-1562387853. [Google Scholar]
- Liu, W.P.; Chen, Y.H.; Ming, X.; Kong, Y. Design and Synthesis of a Novel Cationic Peptide with Potent and Broad-Spectrum Antimicrobial Activity. BioMed Res. Int. 2015, 2015, 578764. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, P.; Singh, M.; Kumari, H.; Kumari, A.; Mukhopadhyay, K. Bactericidal activity of curcumin I is associated with damaging of bacterial membrane. PLoS ONE 2015, 10, e0121313. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Sambrook, J. Isolating DNA from Gram-Negative Bacteria. Cold Spring Harb. Protoc. 2017, 2017, pdb-prot093369. [Google Scholar] [CrossRef]
- Bassetti, M.; Righi, E. Development of novel antibacterial drugs to combat multiple resistant organisms. Langenbecks Arch. Surg. 2015, 400, 153–165. [Google Scholar] [CrossRef]
- Yacoby, I.; Bar, H.; Benhar, I. Targeted drug-carrying bacteriophages as antibacterial nanomedicines. Antimicrob. Agents Chemother. 2007, 51, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnazza, S.; Gioffre, G.; Felici, F.; Guglielmino, S. Recombinant phage probes for Listeria monocytogenes. J. Phys. Condens. Matter 2007, 19, 395011. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Liu, I.J.; Lu, R.M.; Wu, H.C. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.; Wang, C.; Dong, W.; Zhu, W.; Shang, D. Antimicrobial properties and interaction of two Trp-substituted cationic antimicrobial peptides with a lipid bilayer. J. Antibiot. 2014, 67, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial Peptides: Interaction with Model and Biological Membranes and Synergism with Chemical Antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Koh, J.J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Rončević, T.; Puizina, J.; Tossi, A. Antimicrobial Peptides as Anti-Infective Agents in Pre-Post-Antibiotic Era? Int. J. Mol. Sci. 2019, 20, 5713. [Google Scholar] [CrossRef] [Green Version]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirski, T.; Niemcewicz, M.; Bartoszcze, M.; Gryko, R.; Michalski, A. Utilisation of peptides against microbial infections—A review. Ann. Agric. Environ. Med. 2017, 25, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.T.; Espinoza, H.V.; Espinoza, J.L. Emerging superbugs: The threat of Carbapenem Resistant Enterobacteriaceae. AIMS Microbiol. 2020, 6, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Bagheri, M.; Amininasab, M.; Dathe, M. Arginine/Tryptophan-Rich Cyclic α/β-Antimicrobial Peptides: The Roles of Hydrogen Bonding and Hydrophobic/Hydrophilic Solvent-Accessible Surface Areas upon Activity and Membrane Selectivity. Chemistry 2018, 24, 14242–14253. [Google Scholar] [CrossRef]
- Hartmann, M.; Berditsch, M.; Ulrich, A.S. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 2010, 54, 3132–3142. [Google Scholar] [CrossRef] [Green Version]
- Neville, F.; Cahuzac, M.; Gidalevitz, D. Lipid headgroup discrimination by antimicrobial peptide LL-37: Insight into mechanism of action. Biophys. J. 2006, 90, 1275–1287. [Google Scholar] [CrossRef] [Green Version]
- Zweytick, D.; Deutsch, G.; Lohner, K. Studies on lactoferricin-derived Escherichia coli membrane-active peptides reveal differences in the mechanism of N-acylated versus nonacylated peptides. J. Biol. Chem. 2011, 286, 21266–21276. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta 2006, 1758, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Cox, K.H.; Ruiz-Bustos, E.; Courtney, H.S.; Dale, J.B.; Pence, M.A.; Nizet, V.; Aziz, R.K.; Gerling, I.; Price, S.M.; Hasty, D.L. Inactivation of DltA modulates virulence factor expression in Streptococcus pyogenes. PLoS ONE 2009, 4, e5366. [Google Scholar] [CrossRef] [Green Version]
- Torcato, I.M.; Huang, Y.H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.; Troeira Henriques, S. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta 2013, 1828, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, D.; Simerska, P.; Toth, I. Peptides as therapeutics with enhanced bioactivity. Curr. Med. Chem. 2012, 19, 4451–4461. [Google Scholar] [CrossRef]
- Starr, C.G.; Wimley, W.C. Antimicrobial peptides are degraded by the cytosolic proteases of human erythrocytes. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2319–2326. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Wu, Y.; Wang, L.; Ma, C.; Xi, X.; Bininda-Emonds, O.R.P.; Shaw, C.; Chen, T.; Zhou, M. Evaluation of the bioactivity of a mastoparan peptide from wasp venom and of its analogues designed through targeted engineering. Int. J. Biol. Sci. 2018, 14, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, S.A.; Freire, J.M.; Pérez-Peinado, C.; Domingues, M.M.; Gaspar, D.; Vale, N.; Gomes, P.; Andreu, D.; Henriques, S.T.; Castanho, M.A.R.B.; et al. New Potent Membrane-Targeting Antibacterial Peptides from Viral Capsid Proteins. Front. Microbiol. 2017, 8, 775. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.; Piga, K.B.; Hyndman, M.E.; Vogel, H.J. Improving the Activity of Trp-Rich Antimicrobial Peptides by Arg/Lys Substitutions and Changing the Length of Cationic Residues. Biomolecules 2018, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Saiman, L.; Tabibi, S.; Starner, T.D.; San Gabriel, P.; Winokur, P.L.; Jia, H.P.; McCray, P.B., Jr.; Tack, B.F. Cathelicidin peptides inhibit multiply antibiotic-resistant pathogens from patients with cystic fibrosis. Antimicrob. Agents. Chemother. 2001, 45, 2838–2844. [Google Scholar] [CrossRef] [Green Version]
- Yenugu, S.; Hamil, K.G.; French, F.S.; Hall, S.H. Antimicrobial actions of the human epididymis 2 (HE2) protein isoforms, HE2alpha, HE2beta1 and HE2beta2. Reprod. Biol. Endocrinol. 2004, 2, 61. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Wei, R.B.; Luo, H.Y.; Wang, D.F. Isolation and characterization of an antibacterial peptide fraction from the pepsin hydrolysate of half-fin anchovy (Setipinna taty). Molecules 2012, 17, 2980. [Google Scholar] [CrossRef]
- Taute, H.; Bester, M.J.; Neitz, A.W.; Gaspar, A.R. Investigation into the mechanism of action of the antimicrobial peptides Os and Os-C derived from a tick defensin. Peptides 2015, 71, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Wang, K.; Dang, W.; Chen, R.; Xie, J.; Zhang, B.; Song, J.; Wang, R. Two hits are better than one: Membrane-active and DNA binding-related double-action mechanism of NK-18, a novel antimicrobial peptide derived from mammalian NK-lysin. Antimicrob. Agents Chemother. 2013, 57, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, N.; Miller, K.; Hobbs, J.; Rhys-Williams, W.; Love, W.; Chopra, I. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J. Antimicrob. Chemother. 2009, 64, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: The importance of interfacial activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Park, S.C.; Hahm, K.S.; Park, Y. Antimicrobial HPA3NT3 peptide analogs: Placement of aromatic rings and positive charges are key determinants for cell selectivity and mechanism of action. Biochim. Biophys. Acta 2013, 1828, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Su, L.Y.; Willner, D.L.; Segall, A.M. An antimicrobial peptide that targets DNA repair intermediates in vitro inhibits Salmonella growth within murine macrophages. Antimicrob. Agents Chemother. 2010, 54, 1888–1899. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Mojsoska, B.; Jenssen, H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glukhov, E.; Burrows, L.L.; Deber, C.M. Membrane interactions of designed cationic antimicrobial peptides: The two thresholds. Biopolymers 2008, 89, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sani, M.A.; Whitwell, T.C.; Separovic, F. Lipid composition regulates the conformation and insertion of the antimicrobial peptide maculatin 1.1. Biochim. Biophys. Acta 2012, 1818, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöyry, S.; Vattulainen, I. Role of charged lipids in membrane structures—Insight given by simulations. Biochim. Biophys. Acta 2016, 1858, 2322–2333. [Google Scholar] [CrossRef]
- Hädicke, A.; Blume, A. Binding of cationic model peptides (KX)4K to anionic lipid bilayers: Lipid headgroup size influences secondary structure of bound peptides. Biochim. Biophys. Acta Biomembr. 2017, 1859, 415–424. [Google Scholar] [CrossRef]
- Ma, Q.; Jiao, W.; Lv, Y.; Dong, N.; Zhu, X.; Shan, A. Structure-function relationship of Val/Arg-rich peptides: Effects of net charge and pro on activity. Chem. Biol. Drug Des. 2014, 84, 348–353. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Frequency of Selection | Net Charge | Percentage of Hydrophobicity |
|---|---|---|---|---|
| Pa1 | GPVNKSSTILRM | 3/15 | +2 | 33% |
| Pa2 | AHGNAALVARLK | 1/15 | +2 | 58% |
| Pa3 | GLHTSATNLYLH | 2/15 | 0 | 33% |
| Pa4 | KWHWKDKNALRM | 5/15 | +3 | 41% |
| Pa5 | GSLRPGTTNALV | 2/15 | +1 | 33% |
| Pa6 | FGDLTRGQQRGP | 1/15 | +1 | 16% |
| Pa7 | QGTVARLPIFWP | 1/15 | +1 | 50% |
| Bacterial Strains | Relevant Features | MBC (µg/mL) |
|---|---|---|
| P. aeruginosa ATCC 27853 | Reference strain | 8 |
| P. aeruginosa 12594264 | (C.I.) LVXR MXFR DORR ETPR MEMR CAZR CROR FEPR (MDR) | 16 |
| E. coli ATCC 25922 | Reference strain | 8 |
| E. coli 1160702 | C.I. AMCI CFZR CXMR CTXR CROR GENR CIPR (ESBL) | 16 |
| A. baumannii ATCC 19606 | Reference strain | 8 |
| A. junii 1191828 | C.I. CFZR CROR CAZR | 4 |
| K. pneumoniae ATCC 13883 | Reference Strain | 32 |
| K. pneumoniae 1208398 | C.I. AMPR AMCR SAMR TZPR CFZR CXMR FOXR CTXR CAZR CROR FEPR ATMR MEMR AMKR GENR CIPR NITR (CRE) | 8 |
| S. Typhi 1238912 | C.I. CAZR CTXR GENR | 32 |
| S. flexneri 1109563 | C.I. CFXR CFZR CXMR AMKR CIPR | 32 |
| S. marcescens 1191741 | C.I. AMXR CFZR CXMR FOXR | >256 |
| S. aureus ATCC 25923 | Reference strain | 128 |
| E. faecalis ATCC 19433 | Reference strain | >256 |
| S. pyogenes ATCC 19615 | Reference strain | 64 |
| S. anginosus 1360589 | C.I. | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuen, H.L.; Chan, S.Y.; Ding, Y.E.; Lim, S.; Tan, G.C.; Kho, C.L. Development of a Novel Antibacterial Peptide, PAM-5, via Combination of Phage Display Selection and Computer-Assisted Modification. Biomolecules 2023, 13, 466. https://doi.org/10.3390/biom13030466
Yuen HL, Chan SY, Ding YE, Lim S, Tan GC, Kho CL. Development of a Novel Antibacterial Peptide, PAM-5, via Combination of Phage Display Selection and Computer-Assisted Modification. Biomolecules. 2023; 13(3):466. https://doi.org/10.3390/biom13030466
Chicago/Turabian StyleYuen, Hawk Leong, Szn Yi Chan, Yi En Ding, Suxing Lim, Gim Cheong Tan, and Chiew Ling Kho. 2023. "Development of a Novel Antibacterial Peptide, PAM-5, via Combination of Phage Display Selection and Computer-Assisted Modification" Biomolecules 13, no. 3: 466. https://doi.org/10.3390/biom13030466
APA StyleYuen, H. L., Chan, S. Y., Ding, Y. E., Lim, S., Tan, G. C., & Kho, C. L. (2023). Development of a Novel Antibacterial Peptide, PAM-5, via Combination of Phage Display Selection and Computer-Assisted Modification. Biomolecules, 13(3), 466. https://doi.org/10.3390/biom13030466