
Functional Genome Analysis for Immune Cells Provides Clues for Stratification of Systemic Lupus Erythematosus

Abstract
:1. Introduction
2. Associations between SLE Genetic Risk and Immune Cells
2.1. Functions of Disease Susceptibility Polymorphisms
2.2. Functional Genome Database
2.3. Identification of eGenes in Each Immune Cell
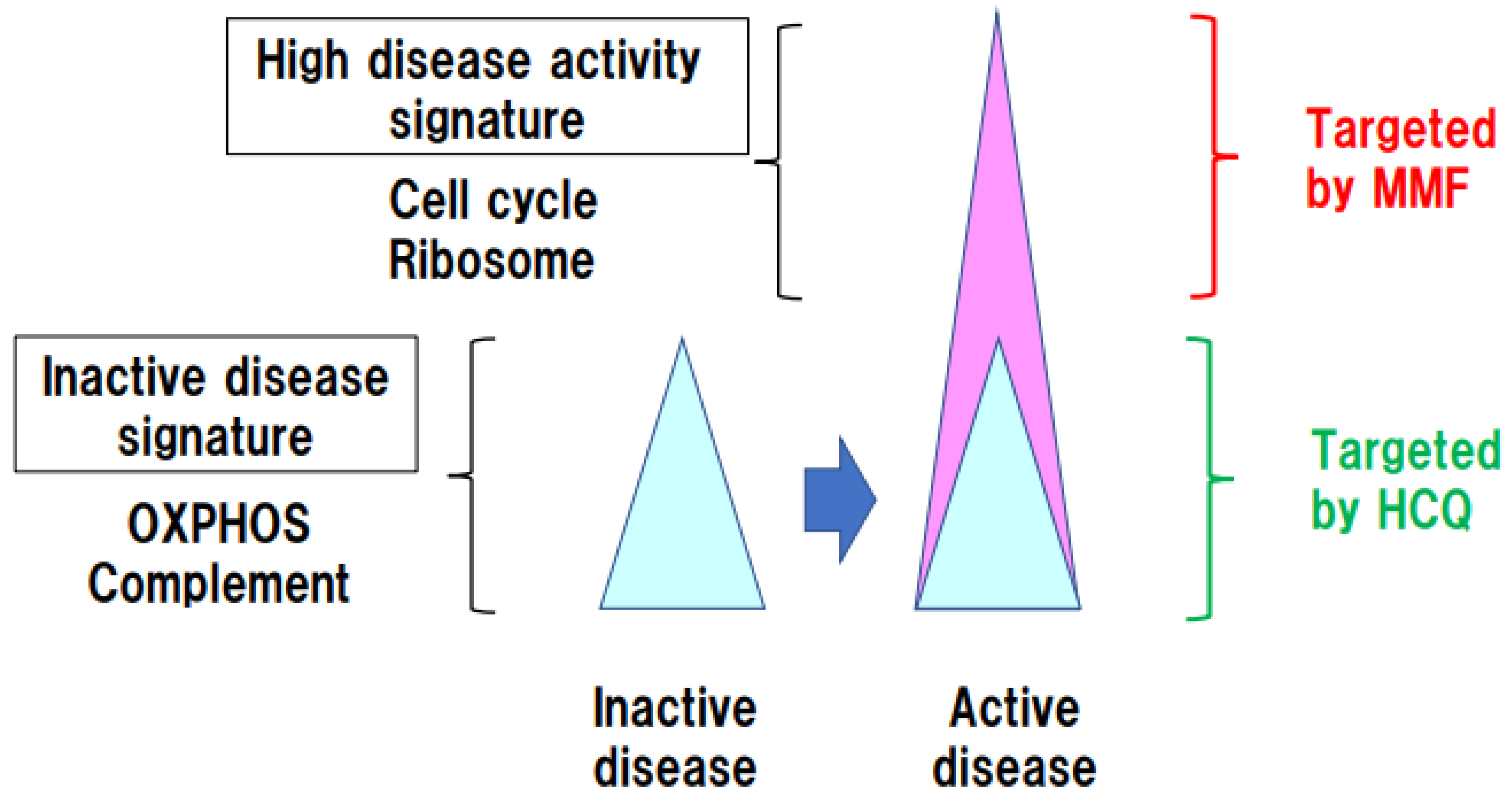
3. Identification of Immune Pathways Associated with Organ Damage in SLE Using Functional Genome Data
4. Persistent OXPHOS Activation in Immune Cells in Inactive SLE Patients
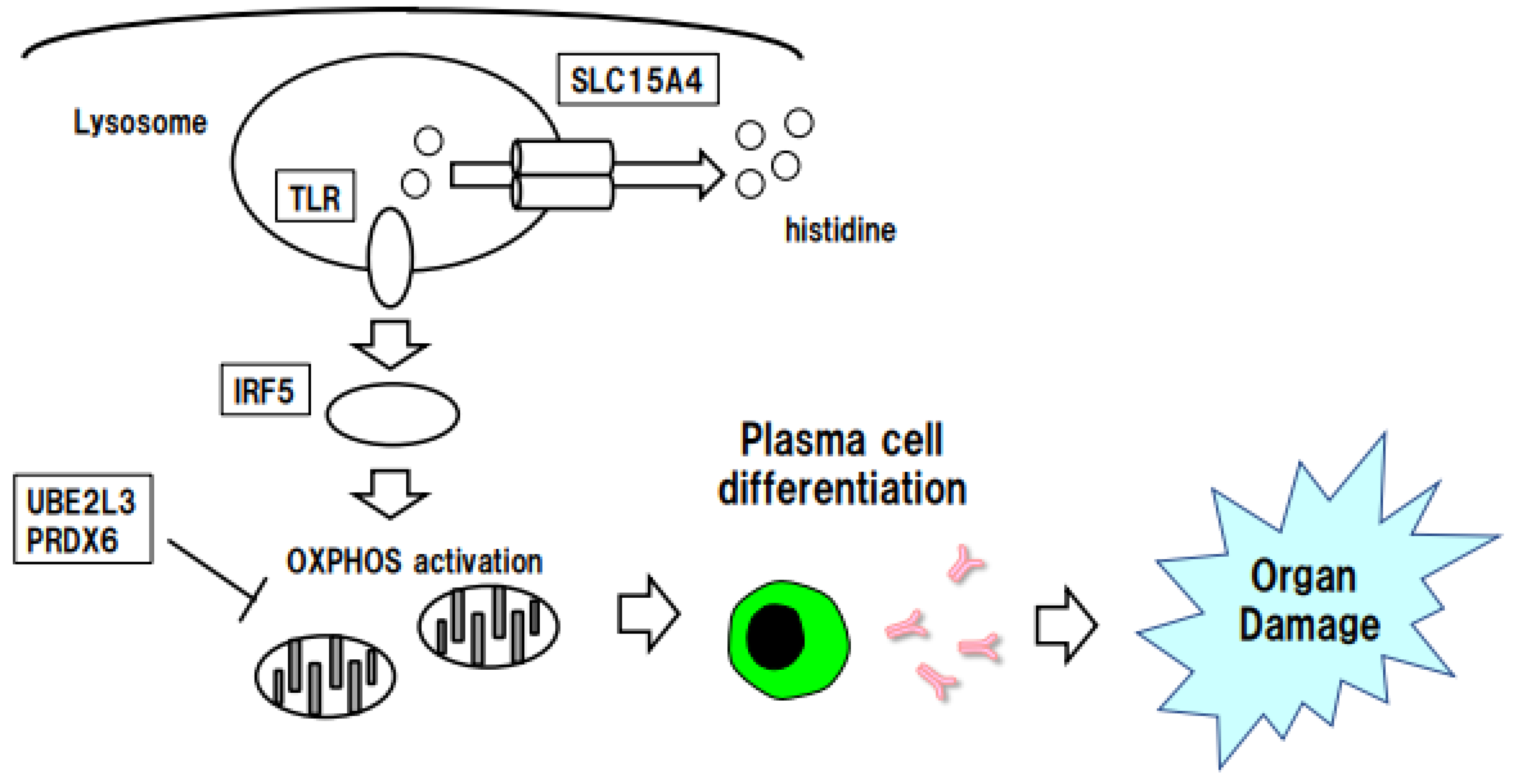
5. Stratification of SLE Risk by SLC15A4-Regulated Histidine
5.1. Function of the SLE eGene SLC15A4
5.2. Histidine Depletion in SLE Plasma and SLC15A4 Function
6. Stratification of SLE Risk According to Immune Cell-Specific IRF5 Expression
6.1. Function of IRF5, an eGene for SLE
6.2. Linkage between IRF5 and OXPHOS
6.3. Potential of IRF5 Gene Polymorphisms in Stratification of SLE Risk
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2016, 2, 16039. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Tziolos, N.; Bertsias, G.; Boumpas, D.T. Update omicronn the diagnosis and management of systemic lupus erythematosus. Ann. Rheum. Dis. 2021, 80, 14–25. [Google Scholar] [CrossRef]
- Thomas, G.; Mancini, J.; Jourde-Chiche, N.; Sarlon, G.; Amoura, Z.; Harle, J.R.; Jougla, E.; Chiche, L. Mortality associated with systemic lupus erythematosus in France assessed by multiple-cause-of-death analysis. Arthritis Rheumatol. 2014, 66, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Bartels, C.M.; Buhr, K.A.; Goldberg, J.W.; Bell, C.L.; Visekruna, M.; Nekkanti, S.; Greenlee, R.T. Mortality and cardiovascular burden of systemic lupus erythematosus in a US population-based cohort. J. Rheumatol. 2014, 41, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Gatto, M.; Zen, M.; Iaccarino, L.; Doria, A. New therapeutic strategies in systemic lupus erythematosus management. Nat. Rev. Rheumatol. 2019, 15, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Mathian, A.; Pha, M.; Haroche, J.; Cohen-Aubart, F.; Hie, M.; Pineton de Chambrun, M.; Boutin, T.H.D.; Miyara, M.; Gorochov, G.; Yssel, H.; et al. Withdrawal of low-dose prednisone in SLE patients with a clinically quiescent disease for more than 1 year: A randomised clinical trial. Ann. Rheum. Dis. 2020, 79, 339–346. [Google Scholar] [CrossRef]
- Elkon, K.B.; Briggs, T.A. The (Orf)ull truth about IRF5 and type I interferons in SLE. Nat. Rev. Rheumatol. 2020, 16, 543–544. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kusuda, M.; Yamaguchi, Y. Interferons and systemic lupus erythematosus: Pathogenesis, clinical features and treatments in interferon-driven disease. Mod. Rheumatol. 2022. ahead of print. [Google Scholar] [CrossRef]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2022, 1–15. [Google Scholar] [CrossRef]
- Bocharnikov, A.V.; Keegan, J.; Wacleche, V.S.; Cao, Y.; Fonseka, C.Y.; Wang, G.; Muise, E.S.; Zhang, K.X.; Arazi, A.; Keras, G.; et al. PD-1hiCXCR5- T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight 2019, 4, e130062. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, Y.; Iwasaki, Y.; Nakano, M.; Narushima, Y.; Ota, M.; Nagafuchi, Y.; Sumitomo, S.; Okamura, T.; Elkon, K.; Ishigaki, K.; et al. Immune cell multiomics analysis reveals contribution of oxidative phosphorylation to B-cell functions and organ damage of lupus. Ann. Rheum. Dis. 2022, 81, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Scherlinger, M.; Tsokos, G.C. Reactive oxygen species: The Yin and Yang in (auto-)immunity. Autoimmun. Rev. 2021, 20, 102869. [Google Scholar] [CrossRef] [PubMed]
- Hisada, R.; Yoshida, N.; Orite, S.Y.K.; Umeda, M.; Burbano, C.; Scherlinger, M.; Kono, M.; Krishfield, S.; Tsokos, G.C. Role of Glutaminase 2 in Promoting CD4+ T Cell Production of Interleukin-2 by Supporting Antioxidant Defense in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2022, 74, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Sul, J.H.; Raj, T.; de Jong, S.; de Bakker, P.I.; Raychaudhuri, S.; Ophoff, R.A.; Stranger, B.E.; Eskin, E.; Han, B. Accurate and fast multiple-testing correction in eQTL studies. Am. J. Hum. Genet. 2015, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Marigorta, U.M.; Denson, L.A.; Hyams, J.S.; Mondal, K.; Prince, J.; Walters, T.D.; Griffiths, A.; Noe, J.D.; Crandall, W.V.; Rosh, J.R.; et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat. Genet. 2017, 49, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic. Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigaki, K.; Kochi, Y.; Suzuki, A.; Tsuchida, Y.; Tsuchiya, H.; Sumitomo, S.; Yamaguchi, K.; Nagafuchi, Y.; Nakachi, S.; Kato, R.; et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat. Genet. 2017, 49, 1120–1125. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Ha, B.; Altay, G.; Greenbaum, J.A.; McVicker, G.; et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 2018, 175, 1701–1715.e16. [Google Scholar] [CrossRef] [Green Version]
- Ota, M.; Nagafuchi, Y.; Hatano, H.; Ishigaki, K.; Terao, C.; Takeshima, Y.; Yanaoka, H.; Kobayashi, S.; Okubo, M.; Shirai, H.; et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 2021, 184, 3006–3021.e17. [Google Scholar] [CrossRef] [PubMed]
- Bentham, J.; Morris, D.L.; Graham, D.S.C.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martin, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Tektonidou, M.G.; Lewandowski, L.B.; Hu, J.; Dasgupta, A.; Ward, M.M. Survival in adults and children with systemic lupus erythematosus: A systematic review and Bayesian meta-analysis of studies from 1950 to 2016. Ann. Rheum. Dis. 2017, 76, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Bultink, I.E.M.; de Vries, F.; van Vollenhoven, R.F.; Lalmohamed, A. Mortality, causes of death and influence of medication use in patients with systemic lupus erythematosus vs matched controls. Rheumatology 2021, 60, 207–216. [Google Scholar] [CrossRef]
- Zen, M.; Iaccarino, L.; Gatto, M.; Bettio, S.; Saccon, F.; Ghirardello, A.; Punzi, L.; Doria, A. The effect of different durations of remission on damage accrual: Results from a prospective monocentric cohort of Caucasian patients. Ann. Rheum. Dis. 2017, 76, 562–565. [Google Scholar] [CrossRef]
- Sumikawa, M.H.; Iwata, S.; Zhang, M.; Miyata, H.; Ueno, M.; Todoroki, Y.; Nagayasu, A.; Kanda, R.; Sonomoto, K.; Torimoto, K.; et al. An enhanced mitochondrial function through glutamine metabolism in plasmablast differentiation in systemic lupus erythematosus. Rheumatology 2022, 61, 3049–3059. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Nguyen-Tien, D.; Ohshima, D.; Karyu, H.; Shimabukuro-Demoto, S.; Yoshida-Sugitani, R.; Toyama-Sorimachi, N. Human SLC15A4 is crucial for TLR-mediated type I interferon production and mitochondrial integrity. Int. Immunol. 2021, 33, 399–406. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Moussaud-Lamodiere, E.L.; Ando, M.; Springer, W. A specific subset of E2 ubiquitin-conjugating enzymes regulate Parkin activation and mitophagy differently. J. Cell Sci. 2014, 127, 3488–3504. [Google Scholar] [CrossRef] [Green Version]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Keung, K.L.; Li, L.; Hu, M.; Lu, B.; Nicholson, L.; Jimenez-Vera, E.; Menon, M.C.; Wei, C.; Alexander, S.; et al. Key driver genes as potential therapeutic targets in renal allograft rejection. JCI Insight 2020, 5, e136220. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Song, Y.; Wang, X.; Sun, J.; Ben, Y.; An, X.; Tong, L.; Bi, J.; Wang, X.; Bai, C. Deletion of peroxiredoxin 6 potentiates lipopolysaccharide-induced acute lung injury in mice. Crit. Care Med. 2011, 39, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, R.K.; Gordon, M.G.; Subramaniam, M.; Kim, M.C.; Hartoularos, G.C.; Targ, S.; Sun, Y.; Ogorodnikov, A.; Bueno, R.; Lu, A.; et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 2022, 376, eabf1970. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Ota, M.; Takeshima, Y.; Iwasaki, Y.; Hatano, H.; Nagafuchi, Y.; Itamiya, T.; Maeda, J.; Yoshida, R.; Yamada, S.; et al. Distinct transcriptome architectures underlying lupus establishment and exacerbation. Cell 2022, 185, 3375–3389.e21. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Shimabukuro-Demoto, S.; Yoshida-Sugitani, R.; Furuyama-Tanaka, K.; Karyu, H.; Sugiura, Y.; Shimizu, Y.; Hosaka, T.; Goto, M.; Kato, N.; et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity 2014, 41, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, Y.; Takeshima, Y.; Nakano, M.; Okubo, M.; Ota, M.; Suzuki, A.; Kochi, Y.; Okamura, T.; Endo, T.; Miki, I.; et al. Combined plasma metabolomic and transcriptomic analysis identify histidine as a biomarker and potential contributor in SLE pathogenesis. Rheumatology 2022, 62, 905–913. [Google Scholar] [CrossRef]
- Graham, R.R.; Kozyrev, S.V.; Baechler, E.C.; Reddy, M.V.; Plenge, R.M.; Bauer, J.W.; Ortmann, W.A.; Koeuth, T.; Gonzalez Escribano, M.F.; Argentine; et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat. Genet. 2006, 38, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.R.; Kyogoku, C.; Sigurdsson, S.; Vlasova, I.A.; Davies, L.R.; Baechler, E.C.; Plenge, R.M.; Koeuth, T.; Ortmann, W.A.; Hom, G.; et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc. Natl. Acad. Sci. USA 2007, 104, 6758–6763. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Kikuchi, M.; Sato, G.R.; Manabe, A.; Tagata, N.; Harita, K.; Nishiyama, A.; Nishimura, K.; Yoshimi, R.; Kirino, Y.; et al. Genetic and chemical inhibition of IRF5 suppresses pre-existing mouse lupus-like disease. Nat. Commun. 2021, 12, 4379. [Google Scholar] [CrossRef]
- Niewold, T.B.; Kelly, J.A.; Kariuki, S.N.; Franek, B.S.; Kumar, A.A.; Kaufman, K.M.; Thomas, K.; Walker, D.; Kamp, S.; Frost, J.M.; et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann. Rheum. Dis. 2012, 71, 463–468. [Google Scholar] [CrossRef]
- Hou, G.; Zhou, T.; Xu, N.; Yin, Z.; Zhu, X.; Zhang, Y.; Cui, Y.; Ma, J.; Tang, Y.; Cheng, Z.; et al. Integrative functional genomics identifies SLE causal genetic variant in the IRF5 risk locus. Arthritis Rheumatol. 2022. ahead of print. [Google Scholar] [CrossRef]
- Heinz, L.X.; Lee, J.; Kapoor, U.; Kartnig, F.; Sedlyarov, V.; Papakostas, K.; Cesar-Razquin, A.; Essletzbichler, P.; Goldmann, U.; Stefanovic, A.; et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature 2020, 581, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Odhams, C.A.; Roberts, A.L.; Vester, S.K.; Duarte, C.S.T.; Beales, C.T.; Clarke, A.J.; Lindinger, S.; Daffern, S.J.; Zito, A.; Chen, L.; et al. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus. Nat. Commun. 2019, 10, 2164. [Google Scholar] [CrossRef] [Green Version]
- Hedl, M.; Yan, J.; Abraham, C. IRF5 and IRF5 Disease-Risk Variants Increase Glycolysis and Human M1 Macrophage Polarization by Regulating Proximal Signaling and Akt2 Activation. Cell Rep. 2016, 16, 2442–2455. [Google Scholar] [CrossRef] [Green Version]
- Albers, G.J.; Iwasaki, J.; McErlean, P.; Ogger, P.P.; Ghai, P.; Khoyratty, T.E.; Udalova, I.A.; Lloyd, C.M.; Byrne, A.J. IRF5 regulates airway macrophage metabolic responses. Clin. Exp. Immunol. 2021, 204, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Matta, B.; Song, S.; Nelson, V.; Diggins, K.; Simpfendorfer, K.R.; Gregersen, P.K.; Linsley, P.; Barnes, B.J. IRF5 genetic risk variants drive myeloid-specific IRF5 hyperactivation and presymptomatic SLE. JCI Insight 2020, 5, e124020. [Google Scholar] [CrossRef] [Green Version]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Nln, I.; Fernandez-Ruiz, R.; Muskardin, T.L.W.; Paredes, J.L.; Blazer, A.D.; Tuminello, S.; Attur, M.; Iturrate, E.; Petrilli, C.M.; Abramson, S.B.; et al. Interferon pathway lupus risk alleles modulate risk of death from acute COVID-19. Transl. Res. 2022, 244, 47–55. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Bentham et al. | Ota et al. | |
|---|---|---|
| 2015 [14] | 2021 [13] | |
| Chr | European | Japanese |
| 1 | FCGR2A | PTPRC |
| FCGR2B | ||
| LYST | ||
| 2 | IFIH1 | LBH |
| 3 | ABHD6 | ARHGAP31 |
| PXK | ||
| 4 | BANK1 | PAQR3 |
| 5 | TCF7 | |
| PTGER4 | ||
| GPX3 | ||
| 6 | UHRF1BP1 | |
| 7 | IRF5 | IRF5 |
| NCF1 | ||
| 8 | BLK | BLK |
| 10 | WDFY4 | |
| 11 | IRF7 | |
| 12 | SLC15A4 | |
| APOLD1 | ||
| 13 | ELF1 | |
| 14 | AHNAK2 | |
| 15 | CSK | RASGRP1 |
| 16 | SOCS1 | IRF8 |
| ITGAM | CDH1 | |
| KAT8 | ||
| 19 | TYK2 | LRRC25 |
| 22 | UBE2L3 | UBE2L3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujio, K. Functional Genome Analysis for Immune Cells Provides Clues for Stratification of Systemic Lupus Erythematosus. Biomolecules 2023, 13, 591. https://doi.org/10.3390/biom13040591
Fujio K. Functional Genome Analysis for Immune Cells Provides Clues for Stratification of Systemic Lupus Erythematosus. Biomolecules. 2023; 13(4):591. https://doi.org/10.3390/biom13040591
Chicago/Turabian StyleFujio, Keishi. 2023. "Functional Genome Analysis for Immune Cells Provides Clues for Stratification of Systemic Lupus Erythematosus" Biomolecules 13, no. 4: 591. https://doi.org/10.3390/biom13040591