Abstract
Insulin resistance is one of the etiologies of type 2 diabetes mellitus (T2DM) and has been suggested to contribute to the development of Alzheimer’s disease by promoting amyloid-β accumulation. Various causes of insulin resistance have been suggested; however, mechanisms of insulin resistance development remain to be elucidated in many respects. Elucidating the mechanisms underlying the development of insulin resistance is one of the key factors in developing methods to prevent the onset of T2DM and Alzheimer’s disease. It has been suggested that the body pH environment plays an important role in the control of cellular functions by regulating the action of hormones including insulin and the activity of enzymes and neurons, thereby maintaining homeostatic conditions of the body. This review introduces: (1) Mitochondrial dysfunction through oxidative stress caused by obesity-induced inflammation. (2) Decreased pH of interstitial fluid due to mitochondrial dysfunction. (3) Development of insulin resistance due to diminution of insulin affinity to its receptor caused by the lowered interstitial fluid pH. (4) Accelerated accumulation of amyloid-β due to elevated activities of β- and γ-secretases caused by the lowered interstitial fluid pH. (5) Diet therapies for improving insulin resistance with weak organic acids that act as bases in the body to raise the pH of lowered interstitial fluid and food factors that promote absorption of weak organic acids in the gut.
1. Introduction
Insulin resistance is characterized by impairment of insulin action in insulin-targeting tissues and organs such as skeletal muscles, adipocytes and the liver, and causes hyperglycemia observed in type 2 diabetes mellitus (T2DM) [1,2]. Continued food intake in excess of requirements causes obesity via hypertrophy and hyperplasia of adipocytes in white adipose tissue (WAT) [3], which is suggested to cause insulin resistance [3]. The hypertrophy of adipocytes produces proinflammatory responses leading to relatively mild chronic inflammation in adipose tissues via physical expansion of adipocyte size [4,5]. This inflammation causes oxidative stress associated with oxidation of DNA, lipids and proteins [6,7,8], which has been suggested to induce insulin resistance via mitochondrial dysfunction [2]. However, it is still unclear how mitochondrial dysfunction causes insulin resistance.
On the one hand, it has been reported that insulin resistance causes the overproduction of free fatty acids, releasing pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and nuclear factor-κB (NF-κB) into the blood circulation [9]. These cytokines further induce activation of serine kinases, IkB kinase (IKK) and c-Jun N-terminal kinase (JNK) [10], thereby inhibiting insulin signaling via the phosphorylation of insulin receptor substrate-1 (IRS-1), blocking activation of insulin receptor substrate (IRS) proteins by binding to the phosphorylated insulin receptor and promoting IRS degradation by ubiquitination [9]. However, it is still unclear if insulin resistance first occurs due to any cause, then induces inflammation via the overproduction of free fatty acids, leading to further dysfunction of insulin signals associated with mitochondrial disorders [9].
Mitochondrial dysfunction [11] results in impaired aerobic glucose metabolism, reducing efficiency of energy (ATP) production [11]. In a state of mitochondrial dysfunction, it becomes necessary to consume more glucose or promote non-glucose metabolism, such as lipids, to maintain ATP production in the amount necessary to sustain vital activities. This situation increases acid production, lowering the pH of the interstitial fluid, which has a pH capacity much smaller than that of blood and the intracellular fluid [12]. As the interstitial fluid pH decreases, the insulin affinity to its receptor is diminished, causing insulin resistance [12].
T2DM patients have an increased risk of developing mental disorders including dementia via neurological dysfunction caused by microvascular complications of diabetes [13]. In addition, patients with T2DM have a higher risk of developing Alzheimer’s disease, another type of dementia [14], due to the development of insulin resistance [14,15,16]. Accumulation of amyloid-β is considered to be one of the main causes of Alzheimer’s disease [17,18], although accumulation of amyloid-β has not yet been confirmed to directly cause Alzheimer’s disease [17,18]. In a state of insulin resistance, the insulin concentration increases. Insulin is a substrate of neprilysin, an amyloid-β-degrading enzyme. Therefore, neprilysin exclusively degrades insulin in the insulin-resistant state, thus reducing the degree of amyloid-β degradation [19]. This leads to accelerated accumulation of amyloid-β in the state of insulin resistance, suggesting that insulin resistance would be a cause of Alzheimer’s disease [15]. Furthermore, acidic conditions elevate the activity of β- and γ-secretases [20,21,22,23,24,25], enzymes that produce amyloid-β from amyloid precursor protein [19,26], resulting in increased amyloid-β production. Thus, the lowered pH of the interstitial fluid observed in insulin resistance accelerates the accumulation of amyloid-β.
Weak organic acids containing carboxyl groups behave as bases inside the body after absorption via Na+-coupled carboxylate transporters (SCT) across the gut [27]. This suggests that intake of weak organic acids improves the lowered interstitial fluid pH via elevation of pH buffer capacity [12,27], leading to amelioration of insulin resistance [12]. In addition, it should be considered that increasing the efficiency of absorption of weak organic acids in the gut is a beneficial way to elevate the lowered interstitial fluid pH.
This narrative review provides evidence for molecular mechanisms regarding development of insulin resistance and comprehensively discusses the following points: (1) Mitochondrial dysfunction due to oxidative stress resulting from obesity-induced inflammation. (2) Lowered interstitial fluid pH due to mitochondrial dysfunction. (3) Development of insulin resistance caused by diminution of insulin affinity to its receptor caused by the lowered interstitial fluid pH. (4) Accelerated accumulation of amyloid-β due to elevated activities of β- and γ-secretases via the lowered interstitial fluid pH. (5) Diet therapies for improving insulin resistance with weak organic acids containing carboxyl groups acting as bases in the body to raise lowered interstitial fluid pH and food factors promoting absorption of weak organic acids in the gut.
To obtain the information on subjects of this review article, I have searched articles providing molecular mechanisms of insulin resistance onset by using the following terms: insulin resistance, interstitial fluid pH, type 2 diabetes mellitus, amyloid-β, Alzheimer’s disease, β- and γ-secretases, weak organic acids, SMCT1 (Na+-coupled monocarboxylate transporter 1), pH capacity, acidosis, ketone bodies, lactic acid and diet therapies in PubMed and Google Scholar.
2. Types of Adipose Tissues and Their Roles in Obesity
Obesity is recognized as a trigger for development of insulin resistance. It is well known that dietary intake in excess of energy expenditure required to maintain vital activities leads to obesity, which is stored in adipose tissues as fat [28,29,30]. Mammals have three types of adipose tissues: (1) white adipose tissue (WAT), (2) brown adipose tissue (BAT) and, (3) beige adipose tissue (BeAT) [28,30]. WAT and BAT are classically recognized, while BeAT has been found recently. WAT, the majority of adipose tissues, contains white adipocytes with unilocular lipid droplets and scarce mitochondria, and plays a role as the primary site of energy (lipid) storage [29]. In contrast, BAT contains brown adipocytes displaying multilocular lipid droplets with a high number of mitochondria, and has thermogenic ability via elevation of uncoupling protein 1 (UCP1) amounts in the inner membrane mitochondria [29]. In BAT, utilization of a high number of mitochondria via elevation of UCP1 leads to uncoupled oxidative phosphorylation from ATP, dissipating chemical energy as heat [29]. Therefore, BAT influences the entire body metabolism, and has the ability to modify the susceptibility to increasing body weight. In humans, BAT was thought to be an energy-producing tissue only in newborns and to regress with age [29]. However, in human adults, BAT has also been identified around the aorta and within the neck supraclavicular region [29]. Furthermore, BeAT, a recently identified type of adipose tissue, is confirmed to have structural and functional features of both WAT and BAT [31,32,33]. BeAT is thought to be generated via conversion of WAT into BeAT and de novo differentiation from a progenitor resident cell [28,29]. Thus, WAT plays a role as the primary site of energy storage, while BAT and BeAT participate in energy expenditure and thermogenesis, although BeAT may also contribute to energy storage. Thus, WAT is more closely linked to the metabolic complications of obesity, such as diabetes.
3. Obesity-Induced Release of Pro-Inflammatory Cytokines and Mitochondrial Damage
Obesity due to continuous dietary intake in excess of energy expenditure is associated with hyperplasia and hypertrophy of adipocytes in WAT [34] and causes excessive release of free fatty acids [35,36]. In the circumstance of adipose tissues, macrophages are polarized into pro-inflammatory M1 macrophages [3], which release pro-inflammatory cytokines, TNF-α and NF-κB [9]. These cytokines activate serine kinases, IKK and JNK [10], which inactivate insulin signals by phosphorylating IRS-1 at serine-302 (pS302) and serine-307 (pS307), instead of its normal tyrosine residue phosphorylation site [9]. IKK and JNK activate further suppressors of cytokine signaling (SOCS) protein, an inflammatory-related negative regulator of IRS proteins, blocking activation of IRS proteins by binding with the phosphorylated insulin receptor and promoting degradation of IRS via ubiquitination [9]. These phenomena induce insulin resistance (Figure 1). Furthermore, immune cells, such as dendritic cells, mast cells, B cells, T cells and neutrophils, appear around WAT under the condition of sustained energy supply in excess of requirements [34]. These phenomena increase levels of proinflammatory cytokines such as interleukin-6 (IL-6), interleukin-1β (IL-1β), chemotactic protein-1 and TNF-α [5] and also produce more oxidative stress [5]. This process further leads to oxidation of DNA, lipids and proteins, which causes mitochondria damage and genetic mutations in DNA/RNA [6] (Figure 1). Derivatives of oxidative reactions are reported to increase in insulin resistance [37]. However, the molecular mechanisms of how mitochondrial damage and DNA/RNA genetic mutations cause the development of insulin resistance remain to be elucidated.
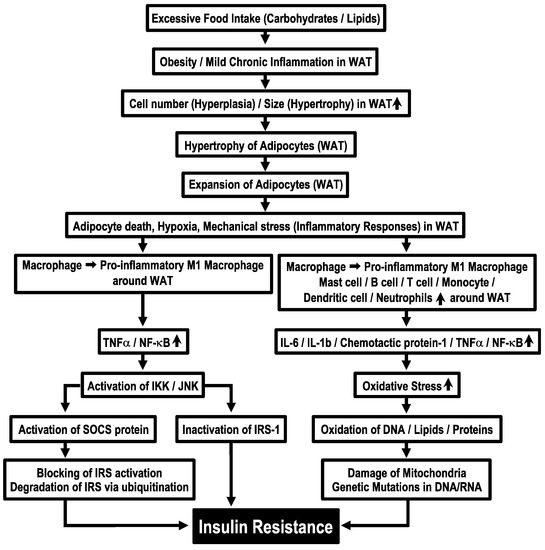
Figure 1.
Molecular mechanisms of obesity-induced insulin resistance. WAT, white adipose tissue.
4. Onset of Insulin Resistance due to Lowered Interstitial Fluid pH Caused by Mitochondrial Damage
As is well known, glucose metabolism is performed via the anaerobic process in the cytoplasm by the glycolytic pathway followed by the aerobic process in mitochondria through the Krebs (TCA) cycle associated with the following two processes: (i) the electron transport chain (ETC) and (ii) the oxidative phosphorylation pathway (OPP; a H+ channel (F0) and ATPase (F1)) [38]. Cells with normal function of mitochondria produce 38 molecules of ATP, 6 molecules of CO2 and 12 molecules of H2O from 1 molecule of glucose, consuming 6 molecules of O2 and 6 molecules of H2O with no net production of H+ under aerobic conditions (Figure 2A) [39,40]. On the one hand, only two molecules of ATP are produced from one molecule of glucose with production of two molecules of lactate– and two equivalents of H+ via the glycolysis process in an anaerobic or mitochondrial dysfunctional state (Figure 2B) [39,40]. Thus, to obtain the ATP needed to sustain vital activity, this state requires the metabolism of far more molecules of glucose than normal mitochondrial function, where O2 is available, producing much more lactate– and H+ [27], which are extruded to the extracellular space (interstitial fluid space) via monocarboxylate transporter (MCT) (Figure 2B). Therefore, it is necessary to consider how the pH condition in a state of mitochondrial dysfunction differs from that in a state of normal mitochondrial function.
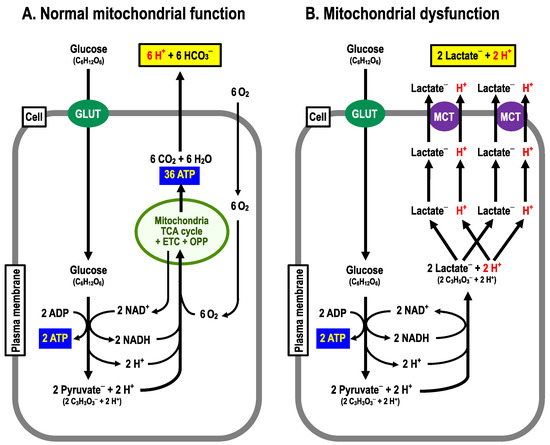
Figure 2.
Metabolizing pathways. (A) Normal mitochondrial function. From 1 molecule of glucose, 38 molecules of ATP, 6 moles of CO2 are produced by consuming 6 molecules of O2. (B) Mitochondrial dysfunction. From one molecule of glucose, two molecules of ATP and two equivalents of H+ are produced.
One pH homeostatic system commonly functioning in the blood, cytosolic space and interstitial fluid is the pH buffering system via bicarbonate-carbonate (HCO3–-CO2)- and phosphoric acid (H3PO4) [41] (Figure 3A). Blood contains H+-binding proteins, such as hemoglobin and albumin, strong pH buffers, keeping blood pH within the narrow normal range (7.35~7.45), and proteins in the intracellular space there also exhibit pH buffering capacity (Figure 3A). In addition, H+ transporters extrude H+ to the extracellular space (the interstitial fluid space) across the plasma membrane. On the one hand, the interstitial fluid has little pH buffer capacity compared with blood and/or the intracellular space, thus the interstitial fluid pH is likely to be variable compared with the pH of blood and/or the intracellular space. In fact, the interstitial fluid pH in T2DM patients with mitochondrial dysfunction is lower than that in healthy individuals (Figure 3B) [11,14,42,43,44,45,46,47,48].
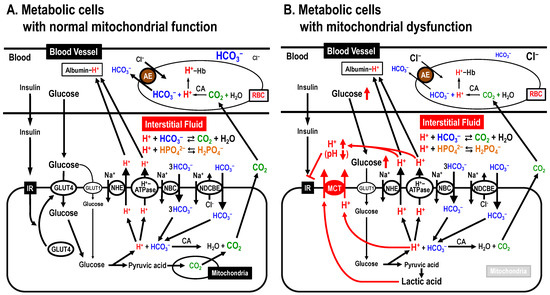
Figure 3.
Mitochondrial dysfunction-induced insulin resistance via lowering interstitial fluid pH. (A) Metabolic cells with normal mitochondrial function. (B) Metabolic cells with mitochondrial dysfunction. AE, anion exchanger; CA, carbonic anhydrase; MCT, monocarboxylate transporter; NBC, Na+-HCO3– cotransporter; NDCBE, Na+-driven Cl–/HCO3– exchanger; NHE, Na+/H+ exchanger. Modified from Figure 3 in Int. J. Mol. Sci. 2018, 19, 3244 ([12]).
The pH of fluids around an enzyme or substrate can alter the binding affinity of the enzyme and substrate, which in turn changes the activity of the enzyme [49]. This is because H+-bindings dramatically change the protein tertiary structure, and H+ also acts on key residues in the catalytic site of the enzyme [49]. pH alteration also changes the binding affinity of insulin and its receptor. Indeed, lowered pH in the interstitial fluid has been reported to diminish the binding affinity of insulin and its receptor, leading to insulin resistance (Figure 3B) [50,51].
Thus, maintenance of the interstitial fluid pH within normal ranges is essential for prevention of pathogenesis of insulin resistance. Strategies for maintenance of the interstitial fluid pH are described later in this article.
5. Insulin Resistance Caused by Lowered pH of the Interstitial Fluid
5.1. Molecular Mechanisms of Co-Occurrence of Insulin Resistance and Chronic Obstructive Pulmonary Disease (COPD) via Lowered pH of the Interstitial Fluid
The CO2 produced in mitochondria of metabolic cells moves into red blood cells (RBC), where CO2 is transformed to H+ and HCO3– via a CA-facilitated process. H+ binds to Hb, while HCO3− is exchanged for extracellular Cl– by an anion exchanger (AE; Cl–/HCO3– exchanger) and excreted out of RBC (Figure 3A). Then, blood moves to the lung via the heart. In the lung, the partial pressure of CO2 is much lower than in environments around peripheral metabolic cells. Therefore, H+ and HCO3– are transformed to CO2 and H2O via a CA-facilitated process in the lung. Then, CO2 moves to the atmosphere via alveolar cells participating in gas exchange (CO2/O2 exchange). COPD patients with disturbances in CO2/O2 exchange in alveolar cells due to alveolar cell damages show high CO2 partial pressure leading to acidic conditions, especially in the interstitial fluid [52]. Multiple cell types including neutrophils in COPD patients produce large amounts of tumor necrosis factor-α (TNF-α) [53], which is one of the factors causing insulin resistance [53]. Thus, COPD patients are at high risk of developing insulin resistance via a TNF-α-mediated pathway [52,54]. However, it should also be considered that the interstitial fluid acidity due to elevated CO2 level in COPD patients [55,56,57,58] would cause insulin resistance [12].
5.2. Molecular Mechanisms of Insulin Resistance Development due to High Salt Intake via Lowered pH of the Interstitial Fluid
High salt (Na+) intake has been reported to be one of the pathogeneses of insulin resistance [59,60,61,62], although little information is available on the molecular mechanism by which high salt intake causes insulin resistance. High salt (Na+) intake leads to an excessive increase in Na+ content in the intravascular and interstitial spaces. High Na+ contents in the interstitial fluid increase the chemical potential of Na+ in the interstitial fluid, resulting in an excess supply of Na+ into the intracellular space. Cells overloaded with Na+ attempt to excrete Na+ out of the cells by Na+,K+-ATPase (Na+,K+-pump), consuming large amounts of ATP. This process lowers the interstitial fluid pH by producing large amounts of H+ [12], leading to insulin resistance [63,64,65,66], although more experimental evidence is required to confirm this process.
5.3. Molecular Mechanisms of Co-Occurrence of Insulin Resistance and Ketone Body/Lactic Acid Production via Lowered pH of the Interstitial Fluid
Ketone bodies, other major H+ sources, are produced from fatty acids under glucose-unavailable pathophysiological conditions in the liver [67] due to insulin resistance and failure of insulin secretion from pancreatic β cells [68,69]. It is notable that even under physiological conditions, ketone bodies are produced by prolonged exercise or extremely low-carbohydrate diet [70,71,72,73]. β-hydroxybutyric acid (β-hydroxybutyrate− + H+) is one of the major ketone bodies [70,71,72,73]. Under glucose-unavailable conditions, β-hydroxybutyric acid serves as an energy source in extrahepatic tissues/organs with normal mitochondrial function [70,71,72,73]. β-hydroxybutyric acid exists as an ionized form, β-hydroxybutyrate− + H+, around normal intracellular pH value, approximately 7.2~7.6, much larger than the pKa (~4.8) of β-hydroxybutyric acid [74].
High production of β-hydroxybutyric acid in hepatocytes leads to lowered intracellular fluid pH even in healthy persons on extremely low-carbohydrate diets or performing prolonged exercise, as well as T2DM patients with mitochondrial dysfunction [71]. This means that extremely low-carbohydrate diets carry a high risk of causing pathologies such as insulin resistance and amyloid-β accumulation (see the description in the following section in detail), therefore we should pay attention to risks generated from extremely low-carbohydrate diets.
Metformin, a biguanide compound, has been developed as a drug inhibiting gluconeogenesis in the liver for reduction of blood glucose levels [75,76,77,78,79,80,81,82,83], since the elevated blood glucose level is one of the most typical symptoms observed in T2DM patients, causing various types of damages including cerebral and myocardial infarctions, and susceptibility to infection [79,84]. Metformin suppresses hepatic gluconeogenesis via inhibition of complex I of the electron transport chain in mitochondria [75,85], resulting in elevation of lactic acid production (Figure 2B) [75,85]. Thus, metformin may induce lactic acidosis [75,85,86,87,88,89], diminishing the insulin action. Imeglimin is also developed for reduction of blood glucose levels: it has a similar structure to metformin, but contains a triazine ring unlike metformin [75,90]. Imeglimin does not cause lactic acidosis unlike metformin, and is characterized by its ability to rebalance respiratory chain activity via partial inhibition of Complex I and correction of deficient Complex III activity, thereby reducing reactive oxygen species (ROS) production and ameliorating insulin resistance [75,90]. In addition, imeglimine is reported to show better glycemic control when used in combination with metformin in T2DM patients [75].
Inhibitors of sodium-dependent glucose transporter 2 (SGLT2), which is involved in sodium-dependent glucose reabsorption in the renal proximal tubules, have also been developed [75,79,82,84]. Although SGLT2-inhibitors are very useful drugs reducing blood glucose levels [75,79], unfortunately some T2DM patients taking SGLT2-inihibitors [79,83,91,92,93,94] show ketoacidosis via generation of ketone bodies such as β-hydroxybutyric acid due to extremely low glucose availability caused by SGLT2-inhibitors [75,79,82,84,95,96], resulting in severe insulin resistance.
6. Accumulation of Amyloid-β Caused by Lowered pH of the Interstitial Fluid in Insulin Resistance
Accumulation of amyloid-β is well known to occur in patients suffering with Alzheimer’s disease [97,98,99,100,101]. Although accumulation of amyloid-β is not yet confirmed to directly cause Alzheimer’s disease [17,18], it is obvious that accumulation of amyloid-β is one of the main causes of Alzheimer’s disease [17,18]. Accumulation of amyloid-β has been also reported to induce hyperphosphorylation of tau protein, causing neural inflammation, neural loss and synaptic impairment associated with cognitive decline and behavioral abnormalities [102].
T2DM patients are at high risk of developing Alzheimer’s disease [103,104,105]. A possible explanation for the higher risk of developing Alzheimer’s disease in T2DM is that: (1) Neprilysin, an enzyme degrading amyloid-β, has another target protein for degradation, insulin; (2) T2DM patients exhibit hyperinsulinemia; (3) in T2DM patients with hyperinsulinemia, neplilysin contributes to degradation of insulin in addition to amyloid-β, resulting in less amyloid-β degradation by neplilysin than in healthy individuals; (4) this process increases the accumulation of amyloid-β [106,107,108]. This explanation is currently well accepted by many medical researchers and physicians. However, activities of β- and γ-secretases, which are involved in the formation of amyloid-β from amyloid precursor protein [19,26], should also be considered. Activities of β- and γ-secretases are known to be elevated by acidic conditions [20,21,22,23,24,25]. Thus, the acidic condition that occurs in T2DM patients increases the production of amyloid-β by elevating activities of β- and γ-secretases [17,18,26,102]. Further, the effects of acidic conditions on the activity of neprilysin, an enzyme degrading amyloid-β, should also be considered [81,100,106,107,109]. It has been reported that neprilysin activity is reduced in acidic environments [110], suggesting that amyloid-β accumulation is enhanced. Thus, the acidic environment observed in T2DM promotes the β- and γ-secretase-facilitated production of amyloid-β and diminishes the neprilysin-facilitated degradation of amyloid-β, leading to elevated amyloid-β accumulation.
We will also consider the relationship between amyloid-β accumulation and high salt intake induced hypertension. Some reports have shown a relationship between salt-sensitive hypertension and Alzheimer’s disease [111,112]. However, little information is available on how these diseases are correlated. One explanation is that the lower interstitial fluid pH observed in salt-sensitive hypertension promotes the accumulation of amyloid-β by increasing the activity of β- and γ-secretases and decreasing the activity of neprilysin [12].
7. Ameliorating Action of Food Compounds on Insulin Resistance and Accumulation of Amyloid-β
Amelioration (elevation) of lowered interstitial pH would be one of the most intrinsically effective therapeutics for insulin resistance in T2DM. A possible method ameliorating the lowered interstitial fluid pH would be intake of weak organic acids [12]. Weak organic acids, such as citric and acetic acids, are tasted as sour substances on the tongue, since they contain H+ [27], while weak organic acids behave as bases in the body after absorption via Na+-coupled carboxylate transporters (SCT) [113,114], which transport only the carboxyl groups but not H+ [113,114]. Therefore, absorbed dietary weak organic acids behave as bases but not acids, elevating the pH buffering capability to ameliorate (elevate) the lowered interstitial fluid pH in T2DM patients. Thus, intake of weak organic acids is a useful way to ameliorate insulin resistance [12].
Furthermore, efficient absorption of dietary weak organic acids should be considered. Brazilian propolis, ninjin’yoeito and mumefural have been reported to ameliorate insulin resistance in T2DM by elevating the lowered interstitial fluid [63,115,116]. Mumefural increases the expression of SMCT1, a carboxylate transporter, in the colon [116], and ninjin’yoeito also tends to have a similar effect [115]. Thus, by increasing the expression of Na+-coupled carboxylate transporters, these compounds elevate the absorbing amount of carboxyl groups, which behave as bases in the body. Therefore, Brazilian propolis, ninjin’yoeito and mumefural ameliorate insulin resistance via an increase in the interstitial fluid pH by elevating the efficiency of carboxyl group absorption. Amelioration of insulin resistance essentially participates in prevention from frailty by stimulating glucose uptake into skeletal muscles [117,118,119,120]. Thus, dietary therapy combining weak organic acids with compounds that enhance the functional expression of weak organic acid transporters is one useful, efficient T2DM treatment strategy that promotes glucose uptake into skeletal muscles, especially for elderly T2DM patients with high risk of frailty. This combining therapy would also be useful in preventing amyloid-β accumulation via decreased activity of β- and γ-secretases and elevated activity of neprilysin.
In addition to ameliorating interstitial fluid pH, ninjin’yoeito is known to improve glucose metabolism in insulin-sensitive tissues via the brainstem Y4 circuit [121]. Brazilian propolis has also been reported to prevent diabetes and obesity by promoting glucagon-like peptide-1 (GLP1) secretion [122]. Further, mumefural has anti-diabetic properties by inhibiting lipogenesis and inducing BAT/BeAT [123]. Thus, Brazilian propolis, ninjin’yoeito and mumefural would ameliorate insulin resistance through coordinated action of the GLP1/brainstem-Y4-circuit/anti-lipogenesis pathways and interstitial fluid pH improvement.
8. Conclusions
The conclusion of the present article is summarized in Figure 4. Interstitial fluid pH is very variable, since the pH-buffering capacity of interstitial fluids is very limited. Because of the limited pH buffering capacity, even if the cytoplasmic space and “arterial” blood are maintained in the normal pH range, the interstitial fluid can easily become acidic if an excess of acidic metabolites are produced in the metabolizing cells. One of the most serious problems caused by lowered interstitial fluid pH is the development of insulin resistance via decreased binding affinity of insulin to its receptor. Another problem caused by lowered interstitial fluid pH would be an increase in accumulation of amyloid-β via elevation of amyloid-β-producing β-/γ-secretase activities and diminution of amyloid-β-degrading neprilysin activity. Intake of foods containing weak organic acids ameliorates (elevates) the lowered interstitial fluid pH in T2DM patients, rescuing T2DM patients from insulin resistance via elevation of insulin binding affinity to its receptor, and ameliorates (diminishes) the accumulation of amyloid-β by regulating activities of β-/γ-secretases and neprilysin. Further, it is essentially important to discover compounds, such as mumefural, that upregulate the expression of Na+-coupled carboxylate transporters involved in the uptake of carboxyl groups contained in weak organic acids via epithelia of the intestine and/or the colon into the body.
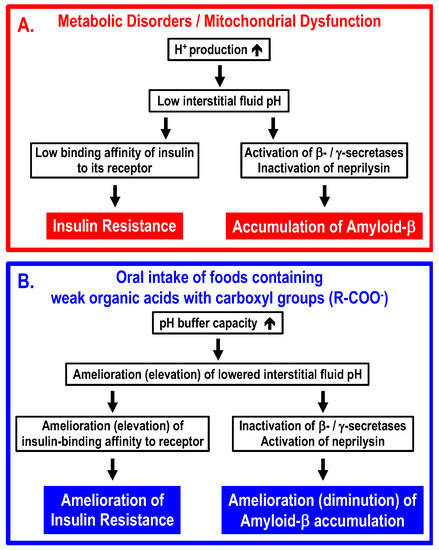
Figure 4.
Molecular mechanisms by which metabolic disorders and/or mitochondrial dysfunction lower interstitial fluid pH, leading to insulin resistance and accumulation of amyloid-β (Panel A), and ameliorating processes by oral intake of foods containing weak organic acids with carboxyl groups (Panel B). (A) Under conditions with metabolic disorders and/or mitochondrial dysfunction, (1) large amounts of protons (H+) are produced in metabolic cells, (2) the large amounts of protons (H+) produced in metabolic cells decrease the interstitial fluid pH due to limited pH-buffering capacity, (3) the lowered interstitial fluid pH diminishes the insulin-binding affinity to its receptor, increases activities of β-/γ-secretases and decreases activities of neprilysin, and (4) the diminution of insulin-binding affinity to its receptor develops insulin resistance, and increased activities of β-/γ-secretases and decreased activity of neprilysin promote accumulation of amyloid-β. (B) Oral intake of foods containing weak organic acids increases the pH capacity of interstitial fluids, ameliorating (elevating) the lowered pH of the interstitial fluid. The amelioration (elevation) of the lowered interstitial fluid pH increases insulin-binding affinity to its receptor, decreases activities of β-/γ-secretases and increases activities of neprilysin, leading to amelioration of insulin resistance and diminution of amyloid-β accumulation.
Funding
This research was funded by Grants-in-Aid for Scientific Research (B) from the Japan Society of the Promotion of Science (JSPS KAKENHI Grant Number JP18H03182 and JP21H03368) to Yoshinori Marunaka.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M. Role of skeletal muscle lipids in the pathogenesis of insulin resistance of obesity and type 2 diabetes. J. Diabetes Investig. 2021, 12, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M.; Deitmer, J.W. Transport metabolons and acid/base balance in tumor cells. Cancers 2020, 12, 899. [Google Scholar] [CrossRef]
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1033–E1045. [Google Scholar] [CrossRef]
- Xu, X.X.; Shi, R.X.; Fu, Y.; Wang, J.L.; Tong, X.; Zhang, S.Q.; Wang, N.; Li, M.X.; Tong, Y.; Wang, W.; et al. Neuronal nitric oxide synthase/reactive oxygen species pathway is involved in apoptosis and pyroptosis in epilepsy. Neural Regen. Res. 2023, 18, 1277–1285. [Google Scholar]
- Lee, S.; Tong, M.; Hang, S.; Deochand, C.; de la Monte, S. CSF and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late Alzheimer’s disease. J. Alzheimers Dis. Park. 2013, 3, 128. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is mitochondrial dysfunction a common root of noncommunicable chronic diseases? Endocr. Rev. 2020, 41, 491–517. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y. The proposal of molecular mechanisms of weak organic acids intake-induced improvement of insulin resistance in diabetes mellitus via elevation of interstitial fluid pH. Int. J. Mol. Sci. 2018, 19, 3244. [Google Scholar] [CrossRef] [PubMed]
- van Sloten, T.T.; Sedaghat, S.; Carnethon, M.R.; Launer, L.J.; Stehouwer, C.D.A. Cerebral microvascular complications of type 2 diabetes: Stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol. 2020, 8, 325–336. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s disease as type 3 dDiabetes: Common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, A.; Torrecilla-Parra, M.; Fernández-de Frutos, M.; Martín-Martín, Y.; Pardo-Marqués, V.; Ramírez, C.M. Posttranscriptional regulation of insulin resistance: Implications for metabolic diseases. Biomolecules 2022, 12, 208. [Google Scholar] [CrossRef] [PubMed]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar]
- Cheng, L.; Li, W.; Chen, Y.; Lin, Y.; Wang, B.; Guo, Q.; Miao, Y. Plasma Aβ as a biomarker for predicting Aβ-PET status in Alzheimer’s disease: A systematic review with meta-analysis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 513–520. [Google Scholar] [CrossRef]
- Sasaguri, H.; Hashimoto, S.; Watamura, N.; Sato, K.; Takamura, R.; Nagata, K.; Tsubuki, S.; Ohshima, T.; Yoshiki, A.; Sato, K.; et al. Recent advances in the modeling of Alzheimer’s disease. Front. Neurosci. 2022, 16, 807473. [Google Scholar] [CrossRef]
- Haass, C.; Hung, A.Y.; Schlossmacher, M.G.; Teplow, D.B.; Selkoe, D.J. beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 1993, 268, 3021–3024. [Google Scholar] [CrossRef]
- Knops, J.; Suomensaari, S.; Lee, M.; McConlogue, L.; Seubert, P.; Sinha, S. Cell-type and amyloid precursor protein-type specific inhibition of A beta release by bafilomycin A1, a selective inhibitor of vacuolar ATPases. J. Biol. Chem. 1995, 270, 2419–2422. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Lou, H.; Ren, X.; Wen, G.; Wu, X.; Xia, X.; Wang, S.; Yu, X.; Yan, L.; Zhang, G.; et al. Ketamine promotes the amyloidogenic pathway by regulating endosomal pH. Toxicology 2022, 471, 153163. [Google Scholar] [CrossRef] [PubMed]
- Ellis, C.R.; Shen, J. pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J. Am. Chem. Soc. 2015, 137, 9543–9546. [Google Scholar] [CrossRef] [PubMed]
- Maesako, M.; Houser, M.C.Q.; Turchyna, Y.; Wolfe, M.S.; Berezovska, O. Presenilin/γ-Secretase Activity Is Located in Acidic Compartments of Live Neurons. J. Neurosci. 2022, 42, 145–154. [Google Scholar] [CrossRef]
- Cai, T.; Hatano, A.; Kanatsu, K.; Tomita, T. Histidine 131 in presenilin 1 is the pH-sensitive residue that causes the increase in Aβ42 level in acidic pH. J. Biochem. 2020, 167, 463–471. [Google Scholar] [CrossRef]
- Hur, J.-Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef]
- Marunaka, Y. Roles of interstitial fluid pH and weak organic acids in development and amelioration of insulin resistance. Biochem. Soc. Trans. 2021, 49, 715–726. [Google Scholar] [CrossRef]
- Bertoncini-Silva, C.; Zingg, J.M.; Fassini, P.G.; Suen, V.M.M. Bioactive dietary components-anti-obesity effects related to energy metabolism and inflammation. Biofactors 2023, 49, 297–321. [Google Scholar] [CrossRef]
- Machado, S.A.; Pasquarelli-do-Nascimento, G.; da Silva, D.S.; Farias, G.R.; de Oliveira Santos, I.; Baptista, L.B.; Magalhães, K.G. Browning of the white adipose tissue regulation: New insights into nutritional and metabolic relevance in health and diseases. Nutr. Metab. 2022, 19, 61. [Google Scholar] [CrossRef]
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: Tracking obesity to its source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef]
- Ishibashi, J.; Seale, P. Medicine. Beige can be slimming. Science 2010, 328, 1113–1114. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, D.; Mehta, R.; Aguilar-Salinas, C.A. Fibroblast Growth Factor 21 and Browning of White Adipose Tissue. Front. Physiol. 2019, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Nunn, E.R.; Shinde, A.B.; Zaganjor, E. Weighing in on idipogenesis. Front. Physiol. 2022, 13, 821278. [Google Scholar] [CrossRef]
- Najjar, S.M.; Abdolahipour, R.; Ghadieh, H.E.; Jahromi, M.S.; Najjar, J.A.; Abuamreh, B.A.M.; Zaidi, S.; Kumarasamy, S.; Muturi, H.T. Regulation of insulin clearance by non-esterified fatty acids. Biomedicines 2022, 10, 1899. [Google Scholar] [CrossRef]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar]
- Thouvenot, K.; Turpin, T.; Taïlé, J.; Clément, K.; Meilhac, O.; Gonthier, M.P. Links between insulin resistance and periodontal bacteria: Insights on molecular players and therapeutic potential of polyphenols. Biomolecules 2022, 12, 378. [Google Scholar] [CrossRef]
- Kamariah, N.; Ragunathan, P.; Shin, J.; Saw, W.G.; Wong, C.F.; Dick, T.; Grüber, G. Unique structural and mechanistic properties of mycobacterial F-ATP synthases: Implications for drug design. Prog. Biophys. Mol. Biol. 2020, 152, 64–73. [Google Scholar] [CrossRef]
- Patel, H.; Kerndt, C.C.; Bhardwaj, A. Physiology, respiratory quotient. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Pessin, J.E.; Bell, G.I. Mammalian facilitative glucose transporter family: Structure and molecular regulation. Annu. Rev. Physiol. 1992, 54, 911–930. [Google Scholar] [CrossRef]
- Lee, D.; Hong, J.H. The Fundamental role of bicarbonate transporters and associated carbonic anhydrase enzymes in maintaining ion and pH homeostasis in non-secretory organs. Int. J. Mol. Sci. 2020, 21, 339. [Google Scholar] [CrossRef]
- Mthembu, S.X.H.; Mazibuko-Mbeje, S.E.; Ziqubu, K.; Nyawo, T.A.; Obonye, N.; Nyambuya, T.M.; Nkambule, B.B.; Silvestri, S.; Tiano, L.; Muller, C.J.F.; et al. Impact of physical exercise and caloric restriction in patients with type 2 diabetes: Skeletal muscle insulin resistance and mitochondrial dysfunction as ideal therapeutic targets. Life Sci. 2022, 297, 120467. [Google Scholar] [CrossRef]
- Shane, M.A.; Nofziger, C.; Blazer-Yost, B.L. Hormonal regulation of the epithelial Na+ channel: From amphibians to mammals. Gen. Comp. Endocrinol. 2006, 147, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Lynn, J.; Park, M.; Ogunwale, C.; Acquaah-Mensah, G.K. A tale of two diseases: Exploring mechanisms linking diabetes mellitus with Alzheimer’s disease. J. Alzheimers Dis. 2022, 85, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Sgarra, L.; Desantis, V.; Nacci, C.; Montagnani, M. Diabetes and Alzheimer’s disease: Might mitochondrial dysfunction help deciphering the common path? Antioxidants 2021, 10, 1257. [Google Scholar] [CrossRef]
- Paul, S.; Saha, D.; Bk, B. Mitochondrial dysfunction and mitophagy closely cooperate in neurological deficits associated with Alzheimer’s disease and type 2 diabetes. Mol. Neurobiol. 2021, 58, 3677–3691. [Google Scholar] [CrossRef]
- Woo, C.Y.; Jang, J.E.; Lee, S.E.; Koh, E.H.; Lee, K.U. Mitochondrial dysfunction in adipocytes as a primary cause of adipose tissue inflammation. Diabetes Metab. J. 2019, 43, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Takano, C.; Ogawa, E.; Hayakawa, S. Insulin resistance in mitochondrial diabetes. Biomolecules 2023, 13, 126. [Google Scholar] [CrossRef]
- Petrenko, A.G.; Zozulya, S.A.; Deyev, I.E.; Eladari, D. Insulin receptor-related receptor as an extracellular pH sensor involved in the regulation of acid-base balance. Biochim. Biophys. Acta 2013, 1834, 2170–2175. [Google Scholar] [CrossRef]
- Hayata, H.; Miyazaki, H.; Niisato, N.; Yokoyama, N.; Marunaka, Y. Lowered extracellular pH is involved in the pathogenesis of skeletal muscle insulin resistance. Biochem. Biophys. Res. Commun. 2014, 445, 170–174. [Google Scholar] [CrossRef]
- Marunaka, Y.; Aoi, W.; Hosogi, S.; Niisato, N.; Yokoyama, N.; Hayata, H.; Miyazaki, H.; Kusuzaki, K.; Taruno, A.; Nomura, T. What is the role of interstitial pH in diabetes mellitus? Improving action of propolis on type Ⅱ diabetes mellitus via pH regulation. Int. J. Mol. Med. 2013, 32 (Suppl. 1), S50. [Google Scholar]
- Agustí, A.; Melén, E.; DeMeo, D.L.; Breyer-Kohansal, R.; Faner, R. Pathogenesis of chronic obstructive pulmonary disease: Understanding the contributions of gene-environment interactions across the lifespan. Lancet Respir. Med. 2022, 10, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.J.; McGettrick, H.M.; Sapey, E. Shared mechanisms of multimorbidity in COPD, atherosclerosis and type-2 diabetes: The neutrophil as a potential inflammatory target. Eur. Respir. Rev. 2020, 29, 190102. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Hobbs, B.D.; Silverman, E.K. Genetics of chronic obstructive pulmonary disease: Understanding the pathobiology and heterogeneity of a complex disorder. Lancet Respir. Med. 2022, 10, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Neder, J.A.; Berton, D.C.; Phillips, D.B.; O’Donnell, D.E. Exertional ventilation/carbon dioxide output relationship in COPD: From physiological mechanisms to clinical applications. Eur. Respir. Rev. 2021, 30, 200190. [Google Scholar] [CrossRef]
- Shigemura, M.; Sznajder, J.I. Elevated CO2 modulates airway contractility. Interface Focus 2021, 11, 20200021. [Google Scholar] [CrossRef]
- Giraud, R.; Banfi, C.; Assouline, B.; De Charrière, A.; Cecconi, M.; Bendjelid, K. The use of extracorporeal CO2 removal in acute respiratory failure. Ann. Intensive Care 2021, 11, 43. [Google Scholar] [CrossRef]
- Caldwell, H.G.; Carr, J.; Minhas, J.S.; Swenson, E.R.; Ainslie, P.N. Acid-base balance and cerebrovascular regulation. J. Physiol. 2021, 599, 5337–5359. [Google Scholar] [CrossRef]
- Fujita, T. The metabolic syndrome in Japan. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5 (Suppl. 1), S15–S18. [Google Scholar] [CrossRef]
- Abdulai, T.; Runqi, T.; Mao, Z.; Oppong, T.B.; Amponsem-Boateng, C.; Wang, Y.; Liu, X.; Zhang, H.; Wang, C. Preference for high dietary salt intake is associated with undiagnosed type 2 diabetes: The henan rural cohort. Front. Nutr. 2020, 7, 537049. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, Y.; Ye, F.; Liu, W.; Jin, X.; Lin, K.; Wang, J.; Gao, Y.; He, L. Effects of probiotics on glycemic control and intestinal dominant flora in patients with type 2 diabetes mellitus: A protocol for systematic review and meta-analysis. Medicine 2020, 99, e23039. [Google Scholar] [CrossRef]
- Aoi, W.; Iwasa, M.; Marunaka, Y. Metabolic functions of flavonoids: From human epidemiology to molecular mechanism. Neuropeptides 2021, 88, 102163. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Hosogi, S.; Niisato, N.; Yokoyama, N.; Hayata, H.; Miyazaki, H.; Kusuzaki, K.; Fukuda, T.; Fukui, M.; Nakamura, N.; et al. Improvement of insulin resistance, blood pressure and interstitial pH in early developmental stage of insulin resistance in OLETF rats by intake of propolis extracts. Biochem. Biophys. Res. Commun. 2013, 432, 650–653. [Google Scholar] [CrossRef]
- Aoi, W.; Marunaka, Y. Importance of pH homeostasis in metabolic health and diseases: Crucial role of membrane proton transport. BioMed Res. Int. 2014, 2014, 598986. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Zou, X.; Xiao, J.B.; Marunaka, Y. Body fluid pH balance in metabolic health and possible benefits of dietary alkaline foods. eFood 2020, 1, 12–23. [Google Scholar] [CrossRef]
- Marunaka, Y.; Niisato, N.; Zou, X.; Xiao, J.B.; Nakahari, T. Food intake targeting and improving acidity in diabetes and cancer. Food Front. 2020, 1, 9–12. [Google Scholar] [CrossRef]
- Fernandes, G.W.; Bocco, B.M.L.C. Hepatic mediators of lipid metabolism and ketogenesis: Focus on fatty liver and diabetes. Curr. Diabetes Rev. 2021, 17, e110320187539. [Google Scholar] [CrossRef] [PubMed]
- Merlotti, D.; Cosso, R.; Eller-Vainicher, C.; Vescini, F.; Chiodini, I.; Gennari, L.; Falchetti, A. Energy metabolism and ketogenic diets: What about the skeletal health? A narrative review and a prospective vision for planning clinical trials on this Issue. Int. J. Mol. Sci. 2021, 22, 435. [Google Scholar] [CrossRef]
- Nasser, S.; Vialichka, V.; Biesiekierska, M.; Balcerczyk, A.; Pirola, L. Effects of ketogenic diet and ketone bodies on the cardiovascular system: Concentration matters. World J. Diabetes 2020, 11, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Dąbek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of cellular biochemistry, epigenetics and metabolomics by ketone bodies. Implications of the ketogenic diet in the physiology of the organism and pathological states. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef]
- Ghimire, P.; Kaul, P.; Dhamoon, A.S. Ketoacidosis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2019. [Google Scholar]
- Newman, J.C.; Verdin, E. ß-hydroxybutyrate: A signaling metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Vincent, J.-L.; Abraham, E.; Kochanek, P.; Moore, F.A.; Kochanek, P.M.; Fink, M.P. Textbook of Critical Care, 7th ed.; Elsevier: Phadelphia, PA, USA, 2017. [Google Scholar]
- Yendapally, R.; Sikazwe, D.; Kim, S.S.; Ramsinghani, S.; Fraser-Spears, R.; Witte, A.P.; La-Viola, B. A review of phenformin, metformin, and imeglimin. Drug Dev. Res. 2020, 81, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.N.; Hussein, U.K.; Yassa, H.D.; Hassan, S.S.; Rashed, L.A. Synergistic actions of vitamin D and metformin on skeletal muscles and insulin resistance of type 2 diabetic rats. J. Cell. Physiol. 2017, 233, 5768–5779. [Google Scholar] [CrossRef] [PubMed]
- Pipeleers, L.; Wissing, K.M.; Hilbrands, R. Acid-base and electrolyte disturbances in patients with diabetes mellitus. Acta Clin. Belg. 2019, 74, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.L.; Martin, A.R.; Kosasih, M.; Caruana, B.T.; Farrell, R. The role of hyperglycemia in endometrial cancer pathogenesis. Cancers 2020, 12, 1191. [Google Scholar] [CrossRef]
- Baumgartner, K.; Devgun, J. Toxicology of medications for diabetes mellitus. Crit. Care Clin. 2021, 37, 577–589. [Google Scholar] [CrossRef]
- Grammatiki, M.; Sagar, R.; Ajjan, R.A. Metformin: Is it still the first line in type 2 diabetes management algorithm? Curr. Pharm. Des. 2021, 27, 1061–1067. [Google Scholar] [CrossRef]
- Inoue, Y.; Masuda, T.; Misumi, Y.; Ando, Y.; Ueda, M. Metformin attenuates vascular pathology by increasing expression of insulin-degrading enzyme in a mixed model of cerebral amyloid angiopathy and type 2 diabetes mellitus. Neurosci. Lett. 2021, 762, 136136. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, S.; Ji, W.; Yao, H.; Lin, L.; Cui, H.; Santos, H.A.; Pan, G. Emerging theranostic nanomaterials in diabetes and its complications. Adv. Sci. 2021, 9, e2102466. [Google Scholar] [CrossRef]
- Sim, R.; Chong, C.W.; Loganadan, N.K.; Fong, A.Y.Y.; Navaravong, L.; Hussein, Z.; Khunti, K.; Lee, S.W.H. Comparative effectiveness of cardiovascular, renal and safety outcomes of second-line antidiabetic drugs use in people with type 2 diabetes: A systematic review and network meta-analysis of randomised controlled trials. Diabet. Med. 2022, 39, e14780. [Google Scholar] [CrossRef]
- Golledge, J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat. Rev. Cardiol. 2022, 19, 456–474. [Google Scholar] [CrossRef] [PubMed]
- Lalau, J.D.; Kajbaf, F.; Protti, A.; Christensen, M.M.; De Broe, M.E.; Wiernsperger, N. Metformin-associated lactic acidosis (MALA): Moving towards a new paradigm. Diabetes Obes. Metab. 2017, 19, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Moser, M.; Prazak, J.; Fuster, D.G.; Schefold, J.C.; Zuercher, P. Metformin’s role in hyperlactatemia and lactic acidosis in ICU patients: A systematic review. Pharmacology 2023, 108, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhu, Y.J.; Zhou, Y.X.; Ding, J.; Liu, J.Y. Metformin in therapeutic applications in human diseases: Its mechanism of action and clinical study. Mol. Biomed. 2022, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Pilot, C.; Marunaka, Y.; Fais, S. Targeting acidity in cancer and diabetes. Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 273–280. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Fleming, G.A.; Chen, K.; Bicsak, T.A. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metabolism 2016, 65, 20–29. [Google Scholar] [CrossRef]
- Hallakou-Bozec, S.; Vial, G.; Kergoat, M.; Fouqueray, P.; Bolze, S.; Borel, A.L.; Fontaine, E.; Moller, D.E. Mechanism of action of Imeglimin: A novel therapeutic agent for type 2 diabetes. Diabetes Obes. Metab. 2021, 23, 664–673. [Google Scholar] [CrossRef]
- Long, B.; Lentz, S.; Koyfman, A.; Gottlieb, M. Euglycemic diabetic ketoacidosis: Etiologies, evaluation, and management. Am. J. Emerg. Med. 2021, 44, 157–160. [Google Scholar] [CrossRef]
- Fathi, A.; Vickneson, K.; Singh, J.S. SGLT2-inhibitors; more than just glycosuria and diuresis. Heart Fail. Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef]
- Kuno, A.; Kimura, Y.; Mizuno, M.; Oshima, H.; Sato, T.; Moniwa, N.; Tanaka, M.; Yano, T.; Tanno, M.; Miki, T.; et al. Empagliflozin attenuates acute kidney injury after myocardial infarction in diabetic rats. Sci. Rep. 2020, 10, 7238. [Google Scholar] [CrossRef]
- Giorgino, F.; Vora, J.; Fenici, P.; Solini, A. Renoprotection with SGLT2 inhibitors in type 2 diabetes over a spectrum of cardiovascular and renal risk. Cardiovasc. Diabetol. 2020, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Delasos, L.; Bazewicz, C.; Sliwinska, A.; Lia, N.L.; Vredenburgh, J. New onset diabetes with ketoacidosis following nivolumab immunotherapy: A case report and review of literature. J. Oncol. Pharm. Pract. 2021, 27, 716–721. [Google Scholar] [CrossRef]
- Locatelli, C.A.A.; Mulvihill, E.E. Islet health, hormone secretion, and insulin responsivity with low-carbohydrate feeding in diabetes. Metabolites 2020, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Tam, K.Y. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2022, 17, 543–549. [Google Scholar] [PubMed]
- Kamble, S.; Barale, S.; Dhanavade, M.; Sonawane, K. Structural significance of neprylysin from Streptococcus suis GZ1 in the degradation of Aβ peptides, a causative agent in Alzheimer’s disease. Comput. Biol. Med. 2021, 136, 104691. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, H.J. Redox-active metal ions and amyloid-degrading enzymes in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 7697. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial dysfunction in Alzheimer’s disease: A biomarker of the future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Kumar, N.; Kumar, V.; Anand, P.; Kumar, V.; Ranjan Dwivedi, A.; Kumar, V. Advancements in the development of multi-target directed ligands for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2022, 61, 116742. [Google Scholar] [CrossRef]
- Peng, X.; Fan, R.; Xie, L.; Shi, X.; Dong, K.; Zhang, S.; Tao, J.; Xu, W.; Ma, D.; Chen, J.; et al. A growing link between circadian rhythms, type 2 diabetes mellitus and Alzheimer’s disease. Int. J. Mol. Sci. 2022, 23, 504. [Google Scholar] [CrossRef]
- Pakdin, M.; Toutounchian, S.; Namazi, S.; Arabpour, Z.; Pouladi, A.; Afsahi, S.; Poudineh, M.; Nasab, M.M.M.; Yaghoobpoor, S.; Deravi, N. Type 2 diabetes mellitus and Alzheimer disease: A review of the potential links. Curr. Diabetes Rev. 2022, 18, e051121197760. [Google Scholar] [PubMed]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin resistance and diabetes mellitus in Alzheimer’s disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef] [PubMed]
- Humpel, C. Intranasal neprilysin rapidly eliminates amyloid-beta plaques, but causes plaque compensations: The explanation why the amyloid-beta cascade may fail? Neural. Regen. Res. 2022, 17, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Ito, Y.; Tanino, T. Effect of high glucose levels on amyloid β production in retinas of spontaneous diabetes mellitus Otsuka Long-Evans Tokushima fatty rats. Biol. Pharm. Bull. 2015, 38, 601–610. [Google Scholar] [CrossRef]
- Lai, M.C.; Liu, W.Y.; Liou, S.S.; Liu, I.M. The citrus flavonoid hesperetin encounters diabetes-mediated Alzheimer-type neuropathologic changes through relieving advanced glycation end-products Inducing endoplasmic reticulum stress. Nutrients 2022, 14, 745. [Google Scholar] [CrossRef]
- Watamura, N.; Kakiya, N.; Nilsson, P.; Tsubuki, S.; Kamano, N.; Takahashi, M.; Hashimoto, S.; Sasaguri, H.; Saito, T.; Saido, T.C. Somatostatin-evoked Aβ catabolism in the brain: Mechanistic involvement of α-endosulfine-K(ATP) channel pathway. Mol. Psychiatry 2022, 27, 1816–1828. [Google Scholar] [CrossRef]
- Miyazaki, H.; Marunaka, Y. Effects of buffer components on pH dependency of recombinant human neprilysin activity. J. Physiol. Sci. 2018, 68 (Suppl. 1), S125. [Google Scholar]
- Gouveia, F.; Camins, A.; Ettcheto, M.; Bicker, J.; Falcão, A.; Cruz, M.T.; Fortuna, A. Targeting brain Renin-Angiotensin System for the prevention and treatment of Alzheimer’s disease: Past, present and future. Ageing Res. Rev. 2022, 77, 101612. [Google Scholar] [CrossRef]
- Souza, L.A.C.; Trebak, F.; Kumar, V.; Satou, R.; Kehoe, P.G.; Yang, W.; Wharton, W.; Feng Earley, Y. Elevated cerebrospinal fluid sodium in hypertensive human subjects with a family history of Alzheimer’s disease. Physiol. Genom. 2020, 52, 133–142. [Google Scholar] [CrossRef]
- Pajor, A.M. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflug. Arch. 2014, 466, 119–130. [Google Scholar] [CrossRef]
- Gomes, S.D.; Oliveira, C.S.; Azevedo-Silva, J.; Casanova, M.R.; Barreto, J.; Pereira, H.; Chaves, S.R.; Rodrigues, L.R.; Casal, M.; Côrte-Real, M.; et al. The role of diet related short-chain fatty acids in colorectal cancer metabolism and survival: Prevention and therapeutic implications. Curr. Med. Chem. 2020, 27, 4087–4108. [Google Scholar] [CrossRef] [PubMed]
- Hosogi, S.; Ohsawa, M.; Kato, I.; Kuwahara, A.; Inui, T.; Inui, A.; Marunaka, Y. Improvement of diabetes mellitus symptoms by intake of ninjin’yoeito. Front. Nutr. 2018, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Hosogi, S.; Kuwahara, A.; Kuwahara, Y.; Tanaka, S.; Shimamoto, C.; Tagawa, N.; Kato, I.; Yoshimoto, K.; Aoi, W.; Takata, K.; et al. Mumefural prevents insulin resistance and amyloid-beta accumulation in the brain by improving lowered interstitial fluid pH in type 2 diabetes mellitus. Biomed. Res. 2023, 44, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Vlachou, E.; Ntikoudi, A.; Owens, D.A.; Nikolakopoulou, M.; Chalimourdas, T.; Cauli, O. Effectiveness of cognitive behavioral therapy-based interventions on psychological symptoms in adults with type 2 diabetes mellitus: An update review of randomized controlled trials. J. Diabetes Complicat. 2022, 36, 108185. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafiz, A.H.; Peters, S.; Sinclair, A.J. Low glycaemic state increases risk of frailty and functional decline in older people with type 2 diabetes mellitus—Evidence from a systematic review. Diabetes Res. Clin. Pract. 2021, 181, 109085. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Sinclair, A.J.; Hill-Briggs, F.; Moran, C.; Biessels, G.J. Type 2 diabetes and cognitive dysfunction-towards effective management of both comorbidities. Lancet Diabetes Endocrinol. 2020, 8, 535–545. [Google Scholar] [CrossRef]
- Uto, N.S.; Amitani, H.; Atobe, Y.; Sameshima, Y.; Sakaki, M.; Rokot, N.; Ataka, K.; Amitani, M.; Inui, A. Herbal medicine Ninjin’yoeito in the treatment of sarcopenia and frailty. Front. Nutr. 2018, 5, 126. [Google Scholar] [CrossRef]
- Zhang, L.; Gopalasingam, G.; Herzog, H. Ninjin’yoeito, a herbal medicine, enhances glucose tolerance in mice. Neuropeptides 2021, 88, 102150. [Google Scholar] [CrossRef]
- Tsuda, T. Anthocyanins and Curcumin: Possible abilities of prevention of diabetes and obesity via stimulation of glucagon-lLike peptide-1 secretion and Induction of beige adipocyte formation. J. Nutr. Sci. Vitaminol. 2022, 68, S110–S112. [Google Scholar] [CrossRef]
- Bu, S.; Yuan, C.; Cao, F.; Xu, Q.; Zhang, Y.; Ju, R.; Chen, L.; Li, Z. Concentrated extract of Prunus mume fruit exerts dual effects in 3T3-L1 adipocytes by inhibiting adipogenesis and inducing beiging/browning. Food Nutr. Res. 2021, 65, 5492. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).