
Evidence of Metabolic Dysfunction in Amyotrophic Lateral Sclerosis (ALS) Patients and Animal Models


Abstract
1. Introduction
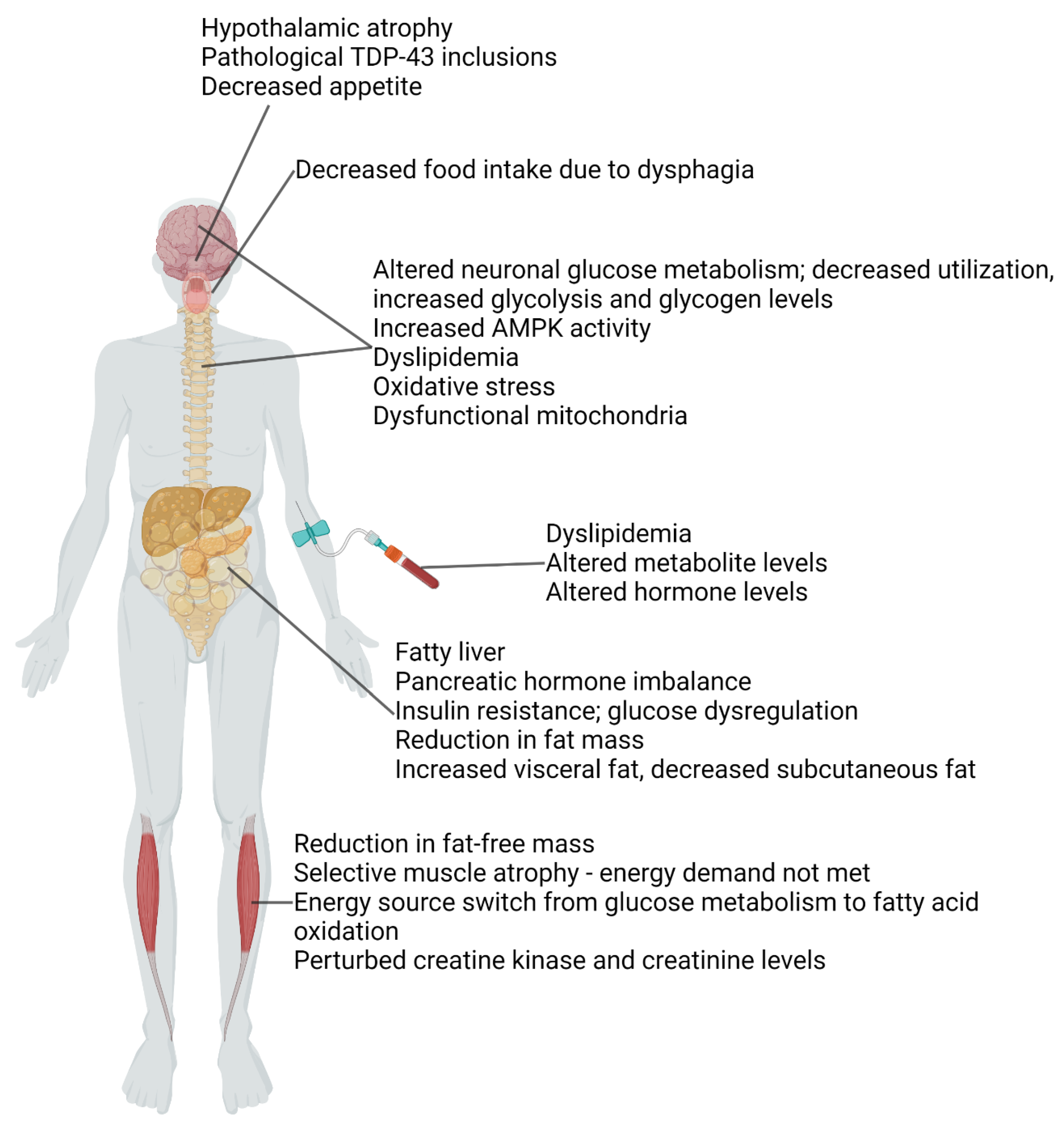
2. Metabolic Dysfunction in ALS Patients and Animal Models
3. Reduced Fat-Free Mass (or Reduced Muscle Mass) and Reduced Glucose Utilization
4. Reduced Fat Mass and Dyslipidemia
5. Liver and Pancreas Dysfunction
6. Metabolic Dysregulation in the Central Nervous System (CNS)
7. Dysfunction of the Hypothalamus, the Hub Regulating Whole-Body Metabolism
8. Treatments Targeting Metabolic Dysfunction
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| BAT | brown adipose tissue |
| BMI | body mass index |
| CK | creatine kinase |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DNA | deoxyribonucleic acid |
| ESC | embryonic stem cell |
| FDA | Food and Drug Administration (federal agency, United States of America) |
| FDG-PET | fluorodeoxyglucose-positron emission tomography |
| GLUT1/3/4 | glucose transporter type 1/3/4 |
| HDL[-C] | high-density lipoprotein (cholesterol) |
| IGF-1 | insulin-like growth factor 1 |
| iPSC | induced pluripotent stem cell |
| LDL[-C] | low-density lipoprotein (cholesterol) |
| MRI | magnetic resonance imaging |
| NMJ | neuromuscular junction |
| REE | resting energy expenditure |
| RNA | ribonucleic acid |
| WAT | white adipose tissue |
References
- Kumar, D.R.; Aslinia, F.; Yale, S.H.; Mazza, J.J. Jean-Martin Charcot: The Father of Neurology. Clin. Med. Res. 2011, 9, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic Lateral Sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Fayemendy, P.; Marin, B.; Labrunie, A.; Boirie, Y.; Walrand, S.; Achamrah, N.; Coëffier, M.; Preux, P.-M.; Lautrette, G.; Desport, J.-C.; et al. Hypermetabolism Is a Reality in Amyotrophic Lateral Sclerosis Compared to Healthy Subjects. J. Neurol. Sci. 2021, 420, 117257. [Google Scholar] [CrossRef] [PubMed]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrère, B.; Couratier, P. Factors Correlated with Hypermetabolism in Patients with Amyotrophic Lateral Sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy Metabolism in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Zucca, I.; Mastropietro, A.; de Rosbo, N.K.; Cavalcante, P.; Tartari, S.; Bonanno, S.; Preite, L.; Mantegazza, R.; Bernasconi, P. Hind Limb Muscle Atrophy Precedes Cerebral Neuronal Degeneration in G93A-SOD1 Mouse Model of Amyotrophic Lateral Sclerosis: A Longitudinal MRI Study. Exp. Neurol. 2011, 231, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Peter, R.S.; Rosenbohm, A.; Dupuis, L.; Brehme, T.; Kassubek, J.; Rothenbacher, D.; Nagel, G.; Ludolph, A.C. Life Course Body Mass Index and Risk and Prognosis of Amyotrophic Lateral Sclerosis: Results from the ALS Registry Swabia. Eur. J. Epidemiol. 2017, 32, 901–908. [Google Scholar] [CrossRef]
- Jawaid, A.; Murthy, S.B.; Wilson, A.M.; Qureshi, S.U.; Amro, M.J.; Wheaton, M.; Simpson, E.; Harati, Y.; Strutt, A.M.; York, M.K.; et al. A Decrease in Body Mass Index Is Associated with Faster Progression of Motor Symptoms and Shorter Survival in ALS. Amyotroph. Lateral Scler. 2010, 11, 542–548. [Google Scholar] [CrossRef]
- Nakayama, Y.; Shimizu, T.; Matsuda, C.; Haraguchi, M.; Hayashi, K.; Bokuda, K.; Nagao, M.; Kawata, A.; Ishikawa-Takata, K.; Isozaki, E. Body Weight Variation Predicts Disease Progression after Invasive Ventilation in Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 12262. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Safer, A.; Wöhrle, J.C.; Palm, F.; Nix, W.A.; Maschke, M.; Grau, A.J. Factors Predicting One-Year Mortality in Amyotrophic Lateral Sclerosis Patients—Data from a Population-Based Registry. BMC Neurol. 2014, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Nakken, O.; Meyer, H.E.; Stigum, H.; Holmøy, T. High BMI Is Associated with Low ALS Risk: A Population-Based Study. Neurology 2019, 93, e424–e432. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Truong, C.T.; Courat, L.; Vallat, J.M.; Couratier, P. Nutritional Assessment and Survival in ALS Patients. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 91–96. [Google Scholar] [CrossRef]
- Holm, T.; Maier, A.; Wicks, P.; Lang, D.; Linke, P.; Münch, C.; Steinfurth, L.; Meyer, R.; Meyer, T. Severe Loss of Appetite in Amyotrophic Lateral Sclerosis Patients: Online Self-Assessment Study. Interact. J. Med. Res. 2013, 2, e8. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Truong, T.C.; Vallat, J.M.; Sautereau, D.; Couratier, P. Nutritional Status Is a Prognostic Factor for Survival in ALS Patients. Neurology 1999, 53, 1059–1063. [Google Scholar] [CrossRef]
- Onesti, E.; Schettino, I.; Gori, M.C.; Frasca, V.; Ceccanti, M.; Cambieri, C.; Ruoppolo, G.; Inghilleri, M. Dysphagia in Amyotrophic Lateral Sclerosis: Impact on Patient Behavior, Diet Adaptation, and Riluzole Management. Front. Neurol. 2017, 8, 94. [Google Scholar] [CrossRef]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS Patients: An Early and Persistent Phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Poehlman, E.T. A Review: Exercise and Its Influence on Resting Energy Metabolism in Man. Med. Sci. Sport. Exerc. 1989, 21, 515. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS Is Associated with Greater Functional Decline and Shorter Survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Vaisman, N.; Lusaus, M.; Nefussy, B.; Niv, E.; Comaneshter, D.; Hallack, R.; Drory, V.E. Do Patients with Amyotrophic Lateral Sclerosis (ALS) Have Increased Energy Needs? J. Neurol. Sci. 2009, 279, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.-M. Body Mass Index, Not Dyslipidemia, Is an Independent Predictor of Survival in Amyotrophic Lateral Sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef]
- Kao, C.S.; van Bruggen, R.; Kim, J.R.; Chen, X.X.L.; Chan, C.; Lee, J.; Cho, W.I.; Zhao, M.; Arndt, C.; Maksimovic, K.; et al. Selective Neuronal Degeneration in MATR3 S85C Knock-in Mouse Model of Early-Stage ALS. Nat. Commun. 2020, 11, 5304. [Google Scholar] [CrossRef]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.; et al. Overexpression of Human Wild-Type FUS Causes Progressive Motor Neuron Degeneration in an Age- and Dose-Dependent Fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef]
- Sephton, C.F.; Tang, A.A.; Kulkarni, A.; West, J.; Brooks, M.; Stubblefield, J.J.; Liu, Y.; Zhang, M.Q.; Green, C.B.; Huber, K.M.; et al. Activity-Dependent FUS Dysregulation Disrupts Synaptic Homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, E4769–E4778. [Google Scholar] [CrossRef]
- Robinson, H.K.; Deykin, A.V.; Bronovitsky, E.V.; Ovchinnikov, R.K.; Ustyugov, A.A.; Shelkovnikova, T.A.; Kukharsky, M.S.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Early Lethality and Neuronal Proteinopathy in Mice Expressing Cytoplasm-Targeted FUS That Lacks the RNA Recognition Motif. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 402–409. [Google Scholar] [CrossRef]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. C9ORF72 Repeat Expansions in Mice Cause TDP-43 Pathology, Neuronal Loss, and Behavioral Deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive Motor Weakness in Transgenic Mice Expressing Human TDP-43. Neurobiol. Dis. 2010, 40, 404–414. [Google Scholar] [CrossRef]
- Cannon, A.; Yang, B.; Knight, J.; Farnham, I.M.; Zhang, Y.; Wuertzer, C.A.; D’Alton, S.; Lin, W.; Castanedes-Casey, M.; Rousseau, L.; et al. Neuronal Sensitivity to TDP-43 Overexpression Is Dependent on Timing of Induction. Acta Neuropathol. 2012, 123, 807–823. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Yang, C.; Qiao, T.; Yu, J.; Wang, H.; Guo, Y.; Salameh, J.; Metterville, J.; Parsi, S.; Yusuf, I.; Brown, R.H.; et al. Low-Level Overexpression of Wild Type TDP-43 Causes Late-Onset, Progressive Neurodegeneration and Paralysis in Mice. PLoS ONE 2022, 17, e0255710. [Google Scholar] [CrossRef]
- Janssens, J.; Wils, H.; Kleinberger, G.; Joris, G.; Cuijt, I.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. Overexpression of ALS-Associated p.M337V Human TDP-43 in Mice Worsens Disease Features Compared to Wild-Type Human TDP-43 Mice. Mol. Neurobiol. 2013, 48, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-F.; Zhang, Y.-J.; Lin, W.-L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of Mutant TDP-43 Induces Neuronal Dysfunction in Transgenic Mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 Mutant Transgenic Mice Develop Features of ALS and Frontotemporal Lobar Degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef]
- Huang, S.-L.; Wu, L.-S.; Lee, M.; Chang, C.-W.; Cheng, W.-C.; Fang, Y.-S.; Chen, Y.-R.; Cheng, P.-L.; Shen, C.-K.J. A Robust TDP-43 Knock-in Mouse Model of ALS. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Jiang, H.; Fu, R.; Zhai, H.; Shi, Y.; Liu, E.; Hirano, M.; Dal Canto, M.C.; Siddique, T. Molecular Dissection of ALS-Associated Toxicity of SOD1 in Transgenic Mice Using an Exon-Fusion Approach. Hum. Mol. Genet. 2008, 17, 2310–2319. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An Adverse Property of a Familial ALS-Linked SOD1 Mutation Causes Motor Neuron Disease Characterized by Vacuolar Degeneration of Mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Steyn, F.J.; Li, R.; Kirk, S.E.; Tefera, T.W.; Xie, T.Y.; Tracey, T.J.; Kelk, D.; Wimberger, E.; Garton, F.C.; Roberts, L.; et al. Altered Skeletal Muscle Glucose–Fatty Acid Flux in Amyotrophic Lateral Sclerosis. Brain Commun. 2020, 2, fcaa154. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; René, F.; de Aguilar, J.-L.G.; Loeffler, J.-P. Evidence for Defective Energy Homeostasis in Amyotrophic Lateral Sclerosis: Benefit of a High-Energy Diet in a Transgenic Mouse Model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, T.; McGoldrick, P.; Fratta, P.; de Oliveira, H.M.; Kent, R.; Phatak, V.; Brandner, S.; Blanco, G.; Greensmith, L.; Acevedo-Arozena, A.; et al. A Nonsense Mutation in Mouse Tardbp Affects TDP43 Alternative Splicing Activity and Causes Limb-Clasping and Body Tone Defects. PLoS ONE 2014, 9, e85962. [Google Scholar] [CrossRef]
- Watanabe, S.; Oiwa, K.; Murata, Y.; Komine, O.; Sobue, A.; Endo, F.; Takahashi, E.; Yamanaka, K. ALS-Linked TDP-43M337V Knock-in Mice Exhibit Splicing Deregulation without Neurodegeneration. Mol. Brain 2020, 13, 8. [Google Scholar] [CrossRef]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. TDP-43 Gains Function Due to Perturbed Autoregulation in a Tardbp Knock-in Mouse Model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.A.; Alix, J.J.P.; Shaw, P.J.; Mead, R.J. Extensive Phenotypic Characterisation of a Human TDP-43Q331K Transgenic Mouse Model of Amyotrophic Lateral Sclerosis (ALS). Sci. Rep. 2021, 11, 16659. [Google Scholar] [CrossRef] [PubMed]
- Stallings, N.R.; Puttaparthi, K.; Dowling, K.J.; Luther, C.M.; Burns, D.K.; Davis, K.; Elliott, J.L. TDP-43, an ALS Linked Protein, Regulates Fat Deposition and Glucose Homeostasis. PLoS ONE 2013, 8, e71793. [Google Scholar] [CrossRef]
- Stephenson, J.; Amor, S. Modelling Amyotrophic Lateral Sclerosis in Mice. Drug Discov. Today Dis. Model. 2017, 25–26, 35–44. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1G93A Mice Predates Disease Onset and Is A Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef] [PubMed]
- Smittkamp, S.E.; Morris, J.K.; Bomhoff, G.L.; Chertoff, M.E.; Geiger, P.C.; Stanford, J.A. SOD1-G93A Mice Exhibit Muscle-Fiber-Type-Specific Decreases in Glucose Uptake in the Absence of Whole-Body Changes in Metabolism. Neurodegener. Dis. 2014, 13, 29–37. [Google Scholar] [CrossRef]
- Tandan, R.; Levy, E.A.; Howard, D.B.; Hiser, J.; Kokinda, N.; Dey, S.; Kasarskis, E.J. Body Composition in Amyotrophic Lateral Sclerosis Subjects and Its Effect on Disease Progression and Survival. Am. J. Clin. Nutr. 2022, 115, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Sun, X.-H.; Cai, Z.-Y.; Shen, D.; Yang, X.-Z.; Liu, M.-S.; Cui, L.-Y. Correlation of Weight and Body Composition with Disease Progression Rate in Patients with Amyotrophic Lateral Sclerosis. Sci. Rep. 2022, 12, 13292. [Google Scholar] [CrossRef]
- Lindauer, E.; Dupuis, L.; Müller, H.-P.; Neumann, H.; Ludolph, A.C.; Kassubek, J. Adipose Tissue Distribution Predicts Survival in Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e67783. [Google Scholar] [CrossRef]
- Goutman, S.A.; Boss, J.; Guo, K.; Alakwaa, F.M.; Patterson, A.; Kim, S.; Savelieff, M.G.; Hur, J.; Feldman, E.L. Untargeted Metabolomics Yields Insight into ALS Disease Mechanisms. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Ingre, C.; Chen, L.; Zhan, Y.; Termorshuizen, J.; Yin, L.; Fang, F. Lipids, Apolipoproteins, and Prognosis of Amyotrophic Lateral Sclerosis. Neurology 2020, 94, e1835–e1844. [Google Scholar] [CrossRef]
- Dorst, J.; Kühnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with Elevated Triglyceride and Cholesterol Serum Levels Have a Prolonged Survival in Amyotrophic Lateral Sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Chełstowska, B.; Barańczyk-Kuźma, A.; Kuźma-Kozakiewicz, M. Dyslipidemia in Patients with Amyotrophic Lateral Sclerosis—A Case Control Retrospective Study. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Larrea, D.; Yun, T.; Xu, Y.; Hupf, J.; Zandkarimi, F.; Chan, R.B.; Mitsumoto, H. Lipidomics Study of Plasma from Patients Suggest That ALS and PLS Are Part of a Continuum of Motor Neuron Disorders. Sci. Rep. 2021, 11, 13562. [Google Scholar] [CrossRef]
- Liu, J.; Luo, X.; Chen, X.; Shang, H. Lipid Profile in Patients With Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Neurol. 2020, 11, 567753. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Lim, Y.S.; Ng, S.-Y.; Ling, S.-C. Deciphering Lipid Dysregulation in ALS: From Mechanisms to Translational Medicine. Transl. Neurodegener. 2022, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Corchuelo, J.M.; Fernández-Beltrán, L.C.; Ali, Z.; Gil-Moreno, M.J.; López-Carbonero, J.I.; Guerrero-Sola, A.; Larrad-Sainz, A.; Matias-Guiu, J.; Matias-Guiu, J.A.; Cunningham, T.J.; et al. Lipid Metabolic Alterations in the ALS-FTD Spectrum of Disorders. Biomedicines 2022, 10, 1105. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.-L.; Sidman, R.L.; et al. Glycosphingolipids Are Modulators of Disease Pathogenesis in Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Kim, H.; Kim, J.-E.; Park, K.S.; Sung, J.-J.; Kim, S.H.; Lee, K.-W. Amyotrophic Lateral Sclerosis Is Associated with Hypolipidemia at the Presymptomatic Stage in Mice. PLoS ONE 2011, 6, e17985. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Priestman, D.A.; Rosenbohm, A.; Dirrig-Grosch, S.; D’Ambra, E.; Huebecker, M.; Hussain, G.; Boursier-Neyret, C.; Echaniz-Laguna, A.; et al. Amyotrophic Lateral Sclerosis and Denervation Alter Sphingolipids and Up-Regulate Glucosylceramide Synthase. Hum. Mol. Genet. 2015, 24, 7390–7405. [Google Scholar] [CrossRef]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Van Den Bosch, L. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef] [PubMed]
- Pagani, M.; Chiò, A.; Valentini, M.C.; Öberg, J.; Nobili, F.; Calvo, A.; Moglia, C.; Bertuzzo, D.; Morbelli, S.; De Carli, F.; et al. Functional Pattern of Brain FDG-PET in Amyotrophic Lateral Sclerosis. Neurology 2014, 83, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Weerasekera, A.; Crabbé, M.; Tomé, S.O.; Gsell, W.; Sima, D.; Casteels, C.; Dresselaers, T.; Deroose, C.; Van Huffel, S.; Rudolf Thal, D.; et al. Non-Invasive Characterization of Amyotrophic Lateral Sclerosis in a HTDP-43A315T Mouse Model: A PET-MR Study. Neuroimage Clin. 2020, 27, 102327. [Google Scholar] [CrossRef]
- Miyazaki, K.; Masamoto, K.; Morimoto, N.; Kurata, T.; Mimoto, T.; Obata, T.; Kanno, I.; Abe, K. Early and Progressive Impairment of Spinal Blood Flow–Glucose Metabolism Coupling in Motor Neuron Degeneration of ALS Model Mice. J. Cereb. Blood Flow Metab. 2012, 32, 456–467. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.-L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.-P.; et al. A Metabolic Switch toward Lipid Use in Glycolytic Muscle Is an Early Pathologic Event in a Mouse Model of Amyotrophic Lateral Sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Browne, S.E.; Yang, L.; DiMauro, J.-P.; Fuller, S.W.; Licata, S.C.; Beal, M.F. Bioenergetic Abnormalities in Discrete Cerebral Motor Pathways Presage Spinal Cord Pathology in the G93A SOD1 Mouse Model of ALS. Neurobiol. Dis. 2006, 22, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of Compromised Blood-Spinal Cord Barrier in Early and Late Symptomatic SOD1 Mice Modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Hirayama, K.; Terao, K. Hepatic Ultrastructural Changes and Liver Dysfunction in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1987, 44, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Nodera, H.; Takamatsu, N.; Muguruma, N.; Ukimoto, K.; Nishio, S.; Oda, M.; Izumi, Y.; Kaji, R. Frequent Hepatic Steatosis in Amyotrophic Lateral Sclerosis: Implication for Systemic Involvement. Neurol. Clin. Neurosci. 2015, 3, 58–62. [Google Scholar] [CrossRef]
- Finkelstein, A.; Kunis, G.; Seksenyan, A.; Ronen, A.; Berkutzki, T.; Azoulay, D.; Koronyo-Hamaoui, M.; Schwartz, M. Abnormal Changes in NKT Cells, the IGF-1 Axis, and Liver Pathology in an Animal Model of ALS. PLoS ONE 2011, 6, e22374. [Google Scholar] [CrossRef]
- Araki, K.; Araki, A.; Honda, D.; Izumoto, T.; Hashizume, A.; Hijikata, Y.; Yamada, S.; Iguchi, Y.; Hara, A.; Ikumi, K.; et al. TDP-43 Regulates Early-Phase Insulin Secretion via CaV1.2-Mediated Exocytosis in Islets. J. Clin. Investig. 2019, 129, 3578–3593. [Google Scholar] [CrossRef]
- McDonald, T.S.; Kumar, V.; Fung, J.N.; Woodruff, T.M.; Lee, J.D. Glucose Clearance and Uptake Is Increased in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis through an Insulin-Independent Mechanism. FASEB J. 2021, 35, e21707. [Google Scholar] [CrossRef]
- Gorges, M.; Vercruysse, P.; Müller, H.-P.; Huppertz, H.-J.; Rosenbohm, A.; Nagel, G.; Weydt, P.; Petersén, Å.; Ludolph, A.C.; Kassubek, J.; et al. Hypothalamic Atrophy Is Related to Body Mass Index and Age at Onset in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1033–1041. [Google Scholar] [CrossRef]
- Liu, S.; Ren, Q.; Gong, G.; Sun, Y.; Zhao, B.; Ma, X.; Zhang, N.; Zhong, S.; Lin, Y.; Wang, W.; et al. Hypothalamic Subregion Abnormalities Are Related to Body Mass Index in Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neurol. 2022, 269, 2980–2988. [Google Scholar] [CrossRef]
- Chang, J.; Shaw, T.B.; Holdom, C.J.; McCombe, P.A.; Henderson, R.D.; Fripp, J.; Barth, M.; Guo, C.C.; Ngo, S.T.; Steyn, F.J.; et al. Lower Hypothalamic Volume with Lower Body Mass Index Is Associated with Shorter Survival in Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2022, 30, 57–68. [Google Scholar] [CrossRef]
- Ye, S.; Luo, Y.; Jin, P.; Wang, Y.; Zhang, N.; Zhang, G.; Chen, L.; Shi, L.; Fan, D. MRI Volumetric Analysis of the Thalamus and Hypothalamus in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 13, 858. [Google Scholar] [CrossRef] [PubMed]
- Bayer, D.; Antonucci, S.; Müller, H.-P.; Saad, R.; Dupuis, L.; Rasche, V.; Böckers, T.M.; Ludolph, A.C.; Kassubek, J.; Roselli, F. Disruption of Orbitofrontal-Hypothalamic Projections in a Murine ALS Model and in Human Patients. Transl. Neurodegener. 2021, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; van Eijk, R.P.A.; Chachay, V.; van den Berg, L.H.; McCombe, P.A.; Henderson, R.D.; Steyn, F.J. Loss of Appetite Is Associated with a Loss of Weight and Fat Mass in Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Vercruysse, P.; Sinniger, J.; El Oussini, H.; Scekic-Zahirovic, J.; Dieterlé, S.; Dengler, R.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; et al. Alterations in the Hypothalamic Melanocortin Pathway in Amyotrophic Lateral Sclerosis. Brain 2016, 139, 1106–1122. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.S.; McCormick, M.C.; Robergs, R.A. Interaction among Skeletal Muscle Metabolic Energy Systems during Intense Exercise. J. Nutr. Metab. 2010, 2010, 905612. [Google Scholar] [CrossRef]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Baxmann, A.C.; Ahmed, M.S.; Marques, N.C.; Menon, V.B.; Pereira, A.B.; Kirsztajn, G.M.; Heilberg, I.P. Influence of Muscle Mass and Physical Activity on Serum and Urinary Creatinine and Serum Cystatin C. Clin. J. Am. Soc. Nephrol. 2008, 3, 348–354. [Google Scholar] [CrossRef]
- Liu, J.; Luo, X.; Chen, X.; Shang, H. Serum Creatinine Levels in Patients with Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 502–508. [Google Scholar] [CrossRef]
- Gao, J.; Dharmadasa, T.; Malaspina, A.; Shaw, P.J.; Talbot, K.; Turner, M.R.; Thompson, A.G. Creatine Kinase and Prognosis in Amyotrophic Lateral Sclerosis: A Literature Review and Multi-Centre Cohort Analysis. J. Neurol. 2022, 269, 5395–5404. [Google Scholar] [CrossRef]
- Chen, X.-P.; Wei, Q.-Q.; Ou, R.-W.; Hou, Y.-B.; Zhang, L.-Y.; Yuan, X.-Q.; Yao, Y.-Q.; Jia, D.-S.; Zhang, Q.; Li, W.-X.; et al. Creatine Kinase in the Diagnosis and Prognostic Prediction of Amyotrophic Lateral Sclerosis: A Retrospective Case-Control Study. Neural Regen. Res. 2021, 16, 591–595. [Google Scholar] [CrossRef]
- Guo, Q.; Hu, W.; Xu, L.; Luo, H.; Wang, N.; Zhang, Q. Decreased Serum Creatinine Levels Predict Short Survival in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, J.; King, R.M.; Jackson, C.E.; Bedlack, R.S.; Barohn, R.J.; Dick, A.; Phillips, L.H.; Chapin, J.; Gelinas, D.F.; Lou, J.-S. Creatine Monohydrate in ALS: Effects on Strength, Fatigue, Respiratory Status and ALSFRS. Amyotroph. Lateral Scler. 2008, 9, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. A Clinical Trial of Creatine in ALS. Neurology 2004, 63, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and Selective Loss of Neuromuscular Synapse Subtypes with Low Sprouting Competence in Motoneuron Diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef]
- Ferri, A.; Coccurello, R. What Is “Hyper” in the ALS Hypermetabolism? Mediat. Inflamm. 2017, 2017, e7821672. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Hegedus, J.; Putman, C.T.; Gordon, T. Time Course of Preferential Motor Unit Loss in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2007, 28, 154–164. [Google Scholar] [CrossRef]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef]
- Hardy, O.T.; Czech, M.P.; Corvera, S. What Causes the Insulin Resistance Underlying Obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 81–87. [Google Scholar] [CrossRef]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose Tissue: Physiology to Metabolic Dysfunction. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Harms, M.; Seale, P. Brown and Beige Fat: Development, Function and Therapeutic Potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.; Tseng, Y.-H. Brown Adipose Tissue. Adipocyte 2012, 1, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Tamucci, K.A.; Namwanje, M.; Fan, L.; Qiang, L. The Dark Side of Browning. Protein Cell 2018, 9, 152–163. [Google Scholar] [CrossRef]
- Ciccarone, F.; Castelli, S.; Lazzarino, G.; Scaricamazza, S.; Mangione, R.; Bernardini, S.; Apolloni, S.; D’Ambrosi, N.; Ferri, A.; Ciriolo, M.R. Lipid Catabolism and Mitochondrial Uncoupling Are Stimulated in Brown Adipose Tissue of Amyotrophic Lateral Sclerosis Mouse Models. Genes Dis. 2022, 10, 321–324. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; Aguilar, J.-L.G.D.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia Is a Protective Factor in Amyotrophic Lateral Sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK Activators: Mechanisms of Action and Physiological Activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and Function of AMPK in Physiology and Diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Scott, J.W.; Kemp, B.E.; Horne, M.K.; Turner, B.J. Mutant TDP-43 Deregulates AMPK Activation by PP2A in ALS Models. PLoS ONE 2014, 9, e90449. [Google Scholar] [CrossRef]
- Lim, M.A.; Selak, M.A.; Xiang, Z.; Krainc, D.; Neve, R.L.; Kraemer, B.C.; Watts, J.L.; Kalb, R.G. Reduced Activity of AMP-Activated Protein Kinase Protects against Genetic Models of Motor Neuron Disease. J. Neurosci. 2012, 32, 1123–1141. [Google Scholar] [CrossRef] [PubMed]
- Pradat, P.-F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.-M.; et al. Impaired Glucose Tolerance in Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2010, 11, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Charchaflie, R.J.; Fernandez, L.B.; Perec, C.J.; Gonzalez, E.; Marzi, A. Functional Studies of the Parotid and Pancreas Glands in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 1974, 37, 863–867. [Google Scholar] [CrossRef]
- Quick, D.T.; Greer, M. Pancreatic Dysfunction in Patients with Amyotrophic Lateral Sclerosis. Neurology 1967, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of Central Nervous System to Body Metabolism in Vertebrates: Its Constancy and Functional Basis. Am. J. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The Remarkable, yet Not Extraordinary, Human Brain as a Scaled-up Primate Brain and Its Associated Cost. Proc. Natl. Acad. Sci. USA 2012, 109, 10661–10668. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical Energy Demands of Signaling and Nonsignaling Components in Brain Are Conserved across Mammalian Species and Activity Levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef]
- Hallermann, S.; de Kock, C.P.J.; Stuart, G.J.; Kole, M.H.P. State and Location Dependence of Action Potential Metabolic Cost in Cortical Pyramidal Neurons. Nat. Neurosci. 2012, 15, 1007–1014. [Google Scholar] [CrossRef]
- Sheng, Z.-H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Sheng, Z.-H. Mechanisms for the Maintenance and Regulation of Axonal Energy Supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the Brain: The Role of Glucose in Physiological and Pathological Brain Function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis Upregulation Is Neuroprotective as a Compensatory Mechanism in ALS. eLife 2019, 8, e45114. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence That Accumulation of Ceramides and Cholesterol Esters Mediates Oxidative Stress-Induced Death of Motor Neurons in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-Chain Fatty Acids, Beta-Hydroxybutyric Acid and Genetic Modulation of the Carnitine Shuttle Are Protective in a Drosophila Model of ALS Based on TDP-43. Front. Mol. Neurosci. 2018, 11, 182. [Google Scholar] [CrossRef]
- Egawa, N.; Izumi, Y.; Suzuki, H.; Tsuge, I.; Fujita, K.; Shimano, H.; Izumikawa, K.; Takahashi, N.; Tsukita, K.; Enami, T.; et al. TDP-43 Regulates Cholesterol Biosynthesis by Inhibiting Sterol Regulatory Element-Binding Protein 2. Sci. Rep. 2022, 12, 7988. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J.J.; Park, N.Y.; Dubey, S.K.; Kim, T.; Ruan, K.; Lim, S.B.; Park, S.-H.; Ha, S.; Kovlyagina, I.; et al. Multi-Omic Analysis of Selectively Vulnerable Motor Neuron Subtypes Implicates Altered Lipid Metabolism in ALS. Nat. Neurosci. 2021, 24, 1673–1685. [Google Scholar] [CrossRef]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn Superoxide Dismutase That Causes Motoneuron Degeneration Is Present in Mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS Interacts with ATP Synthase Beta Subunit and Induces Mitochondrial Unfolded Protein Response in Cellular and Animal Models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis Associated FUS Mutation Shortens Mitochondria and Induces Neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via MPTP to Activate CGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Mórotz, G.M.; Markovinovic, A.; Martín-Guerrero, S.M.; Preza, E.; Arias, N.; Mayl, K.; Aabdien, A.; Gesheva, V.; Nishimura, A.; et al. Disruption of ER-Mitochondria Tethering and Signalling in C9orf72-Associated Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Aging Cell 2022, 21, e13549. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial Bioenergetic Deficits in C9orf72 Amyotrophic Lateral Sclerosis Motor Neurons Cause Dysfunctional Axonal Homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.-D.; Almeida, S.; Gao, F.-B. C9ORF72-ALS/FTD-Associated Poly(GR) Binds Atp5a1 and Compromises Mitochondrial Function in Vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carrì, M.T. Familial ALS-Superoxide Dismutases Associate with Mitochondria and Shift Their Redox Potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of Reduced Glutathione Enhances Motor Neuron Degeneration in Vitro and in Vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 Mutations Causing Amyotrophic Lateral Sclerosis Are Associated with Altered Expression of RNA-Binding Protein HnRNP K and Affect the Nrf2 Antioxidant Pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef]
- Tian, Y.P.; Che, F.Y.; Su, Q.P.; Lu, Y.C.; You, C.P.; Huang, L.M.; Wang, S.G.; Wang, L.; Yu, J.X. Effects of Mutant TDP-43 on the Nrf2/ARE Pathway and Protein Expression of MafK and JDP2 in NSC-34 Cells. Genet. Mol. Res. 2017, 16, gmr16029638. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS Causes DNA Ligation Defects to Inhibit Oxidative Damage Repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef]
- Hall, K.D.; Heymsfield, S.B.; Kemnitz, J.W.; Klein, S.; Schoeller, D.A.; Speakman, J.R. Energy Balance and Its Components: Implications for Body Weight Regulation123. Am. J. Clin. Nutr. 2012, 95, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Cykowski, M.D.; Takei, H.; Schulz, P.E.; Appel, S.H.; Powell, S.Z. TDP-43 Pathology in the Basal Forebrain and Hypothalamus of Patients with Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2014, 2, 171. [Google Scholar] [CrossRef] [PubMed]
- Gabery, S.; Ahmed, R.M.; Caga, J.; Kiernan, M.C.; Halliday, G.M.; Petersén, Å. Loss of the Metabolism and Sleep Regulating Neuronal Populations Expressing Orexin and Oxytocin in the Hypothalamus in Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-World Evidence of Riluzole Effectiveness in Treating Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef]
- Okada, M.; Yamashita, S.; Ueyama, H.; Ishizaki, M.; Maeda, Y.; Ando, Y. Long-Term Effects of Edaravone on Survival of Patients with Amyotrophic Lateral Sclerosis. eNeurologicalSci 2018, 11, 11–14. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; et al. Effect of Sodium Phenylbutyrate/Taurursodiol on Tracheostomy/Ventilation-Free Survival and Hospitalisation in Amyotrophic Lateral Sclerosis: Long-Term Results from the CENTAUR Trial. J. Neurol. Neurosurg. Psychiatry 2022, 93, 871–875. [Google Scholar] [CrossRef]
- Office of the Commissioner FDA Approves New Treatment Option for Patients with ALS. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-option-patients-als (accessed on 24 November 2022).
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A Ketogenic Diet as a Potential Novel Therapeutic Intervention in Amyotrophic Lateral Sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Zhao, Z.; Sui, Y.; Gao, W.; Cai, B.; Fan, D. Effects of Diet on Adenosine Monophosphate-Activated Protein Kinase Activity and Disease Progression in an Amyotrophic Lateral Sclerosis Model. J. Int. Med. Res. 2015, 43, 67–79. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic Triglyceride as a Novel Therapeutic Approach to Effectively Improve the Performance and Attenuate the Symptoms Due to the Motor Neuron Loss in ALS Disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H.M. A High-Fat Jelly Diet Restores Bioenergetic Balance and Extends Lifespan in the Presence of Motor Dysfunction and Lumbar Spinal Cord Motor Neuron Loss in TDP-43A315T Mutant C57BL6/J Mice. Dis. Model. Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef]
- Wills, A.-M.; Hubbard, J.; Macklin, E.A.; Glass, J.; Tandan, R.; Simpson, E.P.; Brooks, B.; Gelinas, D.; Mitsumoto, H.; Mozaffar, T.; et al. Hypercaloric Enteral Nutrition in Amyotrophic Lateral Sclerosis: A Randomized Double-Blind Placebo-Controlled Trial. Lancet 2014, 383, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Dorst, J.; Cypionka, J.; Ludolph, A.C. High-Caloric Food Supplements in the Treatment of Amyotrophic Lateral Sclerosis: A Prospective Interventional Study. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.B.D.C.; Mourão, L.F.; Silva, A.A.; Lima, N.M.F.V.; Almeida, S.R.; Franca, M.C.; Nucci, A.; Amaya-Farfán, J. Effect of Nutritional Supplementation with Milk Whey Proteins in Amyotrophic Lateral Sclerosis Patients. Arq. Neuro-Psiquiatr. 2010, 68, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kihira, T.; Kondo, T.; Kobashi, G.; Washio, M.; Sasaki, S.; Yokoyama, T.; Miyake, Y.; Sakamoto, N.; Inaba, Y.; et al. Nutritional Status and Risk of Amyotrophic Lateral Sclerosis in Japan. Amyotroph. Lateral Scler. 2007, 8, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front. Neurosci. 2019, 13, 532. [Google Scholar] [CrossRef]
- Lalancette-Hebert, M.; Sharma, A.; Lyashchenko, A.K.; Shneider, N.A. Gamma Motor Neurons Survive and Exacerbate Alpha Motor Neuron Degeneration in ALS. Proc. Natl. Acad. Sci. USA 2016, 113, E8316–E8325. [Google Scholar] [CrossRef]


{kind=link}
| Feature | Affected in ALS Patients? | Recapitulated in Mouse Models? |
|---|---|---|
| Reduced body weight | Yes [10,11,13,20,22] | Yes C9ORF72 Tg AAV (GGGGCC)66 [23] C9ORF72 BAC Tg [24] (JAX 029099) FUS WT Tg [25] (JAX 017916) FUS WT Tg [26] (JAX 027898) FUS ΔRRMcyt R522G Tg [27] FUS R521G Tg [26] (JAX 028021) MATR3 S85C KI [28] SOD1 T116X Tg [29] SOD1 G37R Tg [30] (JAX 008229) SOD1 G93A Tg [31] (Gurney et al., JAX 004435) SOD1 G86R Tg [32] (Ripps et al., JAX 005110) TDP-43 WT Tg [33] (JAX 016201) TDP-43 WT Tg [34] TDP-43 WT Tg [35] (JAX 016608) TDP-43 WT Tg [36] (JAX 031609) TDP-43 A315T Tg [33] (JAX 016143) TDP-43 A315T Tg [37] (JAX 010700) TDP-43 M337V Tg [33] TDP-43 M337V Tg [38] TDP-43 M337V Tg [39] (JAX 017604) TDP-43 N390D KI [40] No C9ORF72 BAC Tg [41] (JAX 030222) C9ORF72 BAC Tg [42] JAX (023088, 023099) TDP-43 A315T Tg [43] (Stallings et al.) (increased when compared to control) TDP-43 A315T KI [40] TDP-43 M337V KI [44] TDP-43 Q101X ENU [45] (JAX 019899) TDP-43 Q331K KI [46] (JAX 031345) TDP-43 Q331K Tg [47] (Arnold et al., JAX 017933) (increased when compared to control) See other mutant mice in this review paper [48] |
| Elevated resting energy expenditure | Yes [5,6,21] | Yes SOD1 G93A Tg [31,49] (Gurney et al., JAX 004435) SOD1 G86R Tg [32] (Ripps et al., JAX 005110) No SOD1 G93A Tg [50] (Gurney et al., JAX 002726)TDP-43 A315T Tg [43] (Stallings et al.) (n.d. when normalized to weight) |
| Reduced fat-free mass (or reduced muscle mass) | Yes [20,51,52] | Yes SOD1 G93A Tg [31] (Gurney et al., JAX 004435) TDP-43 A315T Tg [43] (Stallings et al.) TDP-43 Q331K Tg [47] (Arnold et al., JAX 017933) TDP-43 N390D KI [40] No TDP-43 A315T KI [40] |
| Reduced fat mass | Yes [20,51,52] N.d. [53] | Yes SOD1 G86R Tg [32] (Ripps et al., JAX 005110) SOD1 G93A Tg [31] (Gurney et al., JAX 004435) No TDP-43 A315T Tg [43] (Stallings et al.) (increased) |
| Dyslipidemia | Yes [54,55,56,57,58,59] N.d. [60] See more at [61,62] | Yes FUS WT Tg [63] (Mitchell et al., JAX 017916) SOD1 G93A Tg [64] (specific line or JAX not mentioned; B6SJL background) SOD1 G93A Tg [65] (Gurney et al., JAX 002726) SOD1 G86R Tg [66] (Ripps et al., JAX 005110) No TDP-43 A315T Tg [43] (Stallings et al.) |
| Reduced glucose utilization | Yes, in CNS [67] | Yes SOD1 G93A Tg [49,68] in muscles, CNS, respectively (Gurney et al., JAX 004435) SOD1 G86R Tg [69] in muscles (Ripps et al., JAX 005110) SOD1 G93A Tg [70,71] in CNS (Gurney et al., JAX 002726) TDP-43 A315T Tg [43] in muscles (Stallings et al.) TDP-43 A315T Tg [72] in CNS (Wegorzewska et al., JAX 010700) No SOD1 G86R Tg [32] (Ripps et al., JAX 005110) (increased glucose clearance in various tissues, including muscle and CNS) |
| Liver dysfunction | Yes [73,74] | SOD1 G93A Tg [75] (Gurney et al., JAX 002726) |
| Pancreas dysfunction | Yes [76] | SOD1 G93A Tg [77] (Gurney et al., JAX 004435) |
| Degeneration of hypothalamus | Yes [78,79,80] N.d. [81] | Yes SOD1 G93A Tg [82] (Gurney et al., JAX 002726) No FUS ΔNLS KI [82] (Scekic-Zahirovic et al.) |
| Food/caloric intake | Reduced [15,21,83] | Increased FUS ΔNLS KI [84] (Scekic-Zahirovic et al.) (after fasting) SOD1 G86R Tg [32,84] (Ripps et al., JAX 005110) (no fasting; after fasting, respectively) TDP-43 A315T Tg [84] (Wegorzewska et al., JAX 010700) (after fasting) TDP-43 Q331K KI [46] (JAX 031345) SOD1 G93A Tg [32] (Gurney et al., JAX but unclear which background) (n.d.) TDP-43 A315T Tg [43] (Stallings et al.) (n.d. when normalized to weight) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksimovic, K.; Youssef, M.; You, J.; Sung, H.-K.; Park, J. Evidence of Metabolic Dysfunction in Amyotrophic Lateral Sclerosis (ALS) Patients and Animal Models. Biomolecules 2023, 13, 863. https://doi.org/10.3390/biom13050863
Maksimovic K, Youssef M, You J, Sung H-K, Park J. Evidence of Metabolic Dysfunction in Amyotrophic Lateral Sclerosis (ALS) Patients and Animal Models. Biomolecules. 2023; 13(5):863. https://doi.org/10.3390/biom13050863
Chicago/Turabian StyleMaksimovic, Katarina, Mohieldin Youssef, Justin You, Hoon-Ki Sung, and Jeehye Park. 2023. "Evidence of Metabolic Dysfunction in Amyotrophic Lateral Sclerosis (ALS) Patients and Animal Models" Biomolecules 13, no. 5: 863. https://doi.org/10.3390/biom13050863
APA StyleMaksimovic, K., Youssef, M., You, J., Sung, H.-K., & Park, J. (2023). Evidence of Metabolic Dysfunction in Amyotrophic Lateral Sclerosis (ALS) Patients and Animal Models. Biomolecules, 13(5), 863. https://doi.org/10.3390/biom13050863