


Mathematical and Machine Learning Approaches for Classification of Protein Secondary Structure Elements from Cα Coordinates


Abstract
:1. Introduction
- In a crystal, atoms and molecules arrange themselves in regular arrays and X-ray crystallography technology, which has been in use since the 1950s, utilizes this fact to generate atomic and molecular structure of the crystal. In order to determine the atomic structure of a protein, it first needs to be crystallized. However, protein crystallization is a difficult process and not possible for all proteins. For example, outer membrane proteins, mostly -Barrel architectures, of Gram negative bacteria are mostly rigid and stable and therefore X-ray crystallography can be applied relatively easily to determine their molecular structures. However, high-resolution diffracting crystals of plasma membrane proteins and large molecules are not easy to crystallize, due to difficulty of obtaining homogeneous protein samples.
- NMR spectroscopy employs the properties of nuclear spin in the presence of an applied magnetic field to analyze the alignment of atoms’ nuclei and it also provides information about dynamic molecular interactions. NMR spectroscopy requires a large amount of pure samples and as with X-ray crystallography, it has difficulty analyzing molecules with large molecular weight.
- Cryo-EM provides a lower resolution view of a protein compared to X-ray crystallography. However, it does not require crystallization and therefore many proteins that are difficult to crystallize and large protein assemblies can be imaged using Cryo-EM. It creates a 3D image using thousands of 2D projections. Cryo-EM provides different level of views at near-atomic (<5 Å), subnanometer (5–10 Å), and nanometer (>10 Å), resolutions. Only near-atomic resolution can be used to identify locations of and other atoms in the backbone of a protein chain.
2. Materials and Methods
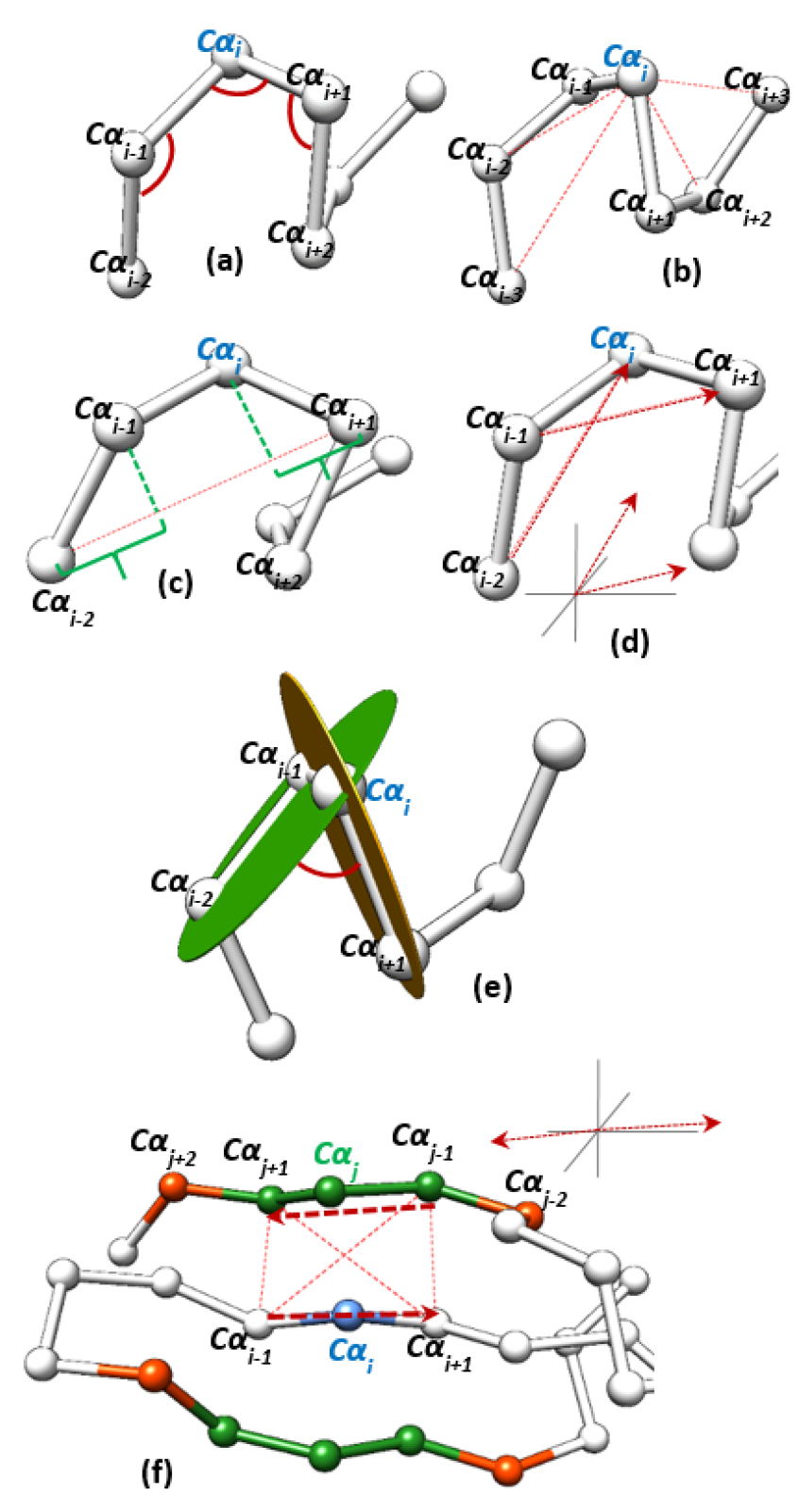
2.1. Feature Generation
2.1.1. Geometric Features
2.1.2. Determining Relevant Features
- 1.
- Determine , the number of most relevant features.
- 2.
- Determine those k features.
| Algorithm 1: Reduction of features. |
| Require: d × N feature matrix F. |
| 1: Estimate effective-rank k of (using rank estimation technique in [40]). |
| 2: Find a sub matrix with k rows and call it . |
| 3: while effective- do |
| 4: Find another sub matrix with k rows and call it . |
| 5: end while |
| 6: k features corresponding to each row of are most relevant features. |
2.2. Mathematical Approach
2.2.1. SSE Subspace Modeling
| Algorithm 2: SSE subspace matching. |
| Require: q: window size, z: window-sliding size, : the number of amino acids. |
| 1: Create empty data matrices , , and . |
| 2: Form first possible window. |
| 3: for all do |
| 4: Form column vector . |
| 5: Expand the corresponding data matrix by adding as a new column vector. |
| 6: Slide the window by z. |
| 7: end for |
| 8: for all data matrices do |
| 9: Compute SVD. For example, . |
| 10: Estimate the rank (using rank estimation technique in [40]). For example, . |
| 11: Compute a subspace. For example, , i.e., the span of the first columns of . |
| 12: end for |
2.2.2. Projections on SSE Subspaces and Classification
- Let be with truncation after the first columns.
- Let be the data vector for the that is being classified. Note that, the same window size as in the training is used.
2.2.3. Local Subspaces and Classification
- A group of neighbors of is identified as illustrated in Figure 2. Each atom in the neighborhood is in using the same window size as before.
- Let be the number of atoms in each neighborhood.
- Construct matrix whose columns are representation of each in the neighborhood. Call this matrix .
- Compute SVD of .
- Let be the local subspace spanned by the columns of .
2.2.4. Post Processing
2.3. Deep Learning Approach
2.3.1. Dataset
2.3.2. Network Architecture and Training Parameters
2.3.3. Evaluation Measures
2.3.4. Joint Prediction of Multiple Architectures
2.4. Ensemble of Machine Learning Models Approach (EML)
3. Results
3.1. Results: Subspace Segmentation Approach
3.2. Results: Deep Learning Approach
3.3. Results: Ensemble of Machine Learning Models
3.4. Results: Existing Approach
3.5. Results: Summary
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ridley, M. Genome, 1st ed.; Harper Perennial: New York, NY, USA, 2000; p. 352. [Google Scholar]
- Murray, R.K.; Granner, D.K.; Mayes, P.A.; Rodwell, V.W. Harper’s Illustrated Biochemistry; McGraw-Hill Medical: Irvine, CA, USA, 2006. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Tarry, M.J.; Haque, A.S.; Bui, K.H.; Schmeing, T.M. X-Ray Crystallography and Electron Microscopy of Cross- and Multi-Module Nonribosomal Peptide Synthetase Proteins Reveal a Flexible Architecture. Structure 2017, 25, 783–793.e4. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Schertler, G.F.X. Membrane Protein Crystallization; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2020; pp. 187–210. [Google Scholar]
- Maveyraud, L.; Mourey, L. Protein X-ray Crystallography and Drug Discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef] [PubMed]
- Hatzakis, E. Nuclear Magnetic Resonance (NMR) Spectroscopy in Food Science: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Y.; Skolnick, J. Application of sparse NMR restraints to large-scale protein structure prediction. Biophys J. 2004, 87, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Danev, R.; Yanagisawa, H.; Kikkawa, M. Cryo-Electron Microscopy Methodology: Current Aspects and Future Directions. Trends Biochem. Sci. 2019, 44, 837–848. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260. [Google Scholar] [CrossRef]
- Terashi, G.; Kihara, D. De novo main-chain modeling for EM maps using MAINMAST. Nat. Commun. 2018, 9, 1618. [Google Scholar] [CrossRef]
- Chen, M.; Baldwin, P.R.; Ludtke, S.J.; Baker, M.L. De Novo modeling in cryo-EM density maps with Pathwalking. J. Struct. Biol. 2016, 196, 289–298. [Google Scholar] [CrossRef]
- Al Nasr, K.; Chen, L.; Si, D.; Ranjan, D.; Zubair, M.; He, J. Building the Initial Chain of the Proteins through de Novo Modeling of the Cryo-Electron Microscopy Volume Data at the Medium Resolutions. In Proceedings of the BCB ’12 ACM Conference on Bioinformatics, Computational Biology and Biomedicine, New York, NY, USA, 7–10 October 2012; pp. 490–497. [Google Scholar] [CrossRef]
- Al Nasr, K. De Novo Protein Structure Modeling from Cryoem Data through a Dynamic Programming Algorithm in the Secondary Structure Topology Graph. Ph.D. Dissertation, Old Dominion University, Norfolk, VA, USA, 2012. [Google Scholar]
- Al Nasr, K.; He, J. Constrained cyclic coordinate descent for cryo-EM images at medium resolutions: Beyond the protein loop closure problem. Robotica 2016, 34, 1777–1790. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Hassabis, D.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Pakhrin, S.C.; Shrestha, B.; Adhikari, B.; Kc, D.B. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci. 2021, 22, 5553. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.D.; Das, S.; Sillitoe, I.; Orengo, C. An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallogr. Sect. D 2017, 73, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.B.; Zhang, Y.; Skolnick, J. TASSER-Lite: An automated tool for protein comparative modeling. Biophys J. 2006, 91, 4180–4190. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.J. Methods to Estimate the Conformation of Proteins and Polypeptides from Circular Dichroism Data. Anal. Biochem. 1996, 235, 1–10. [Google Scholar] [CrossRef]
- Provencher, S.W.; Gloeckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef]
- Dousseau, F.; Pezolet, M. Determination of the secondary structure content of proteins in aqueous solutions from their amide I and amide II infrared bands. Comparison between classical and partial least-squares methods. Biochemistry 1990, 29, 8771–8779. [Google Scholar] [CrossRef]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef]
- Pastore, A.; Saudek, V. The relationship between chemical shift and secondary structure in proteins. J. Magn. Reson. 1990, 90, 165–176. [Google Scholar] [CrossRef]
- Law, S.M.; Frank, A.T.; Brooks, C.L., III. PCASSO: A fast and efficient Cα-based method for accurately assigning protein secondary structure elements. J. Comput. Chem. 2014, 35, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Greer, J. Automatic identification of secondary structure in globular proteins. J. Mol. Biol. 1977, 114, 181–239. [Google Scholar] [CrossRef]
- Richards, F.M.; Kundrot, C.E. Identification of structural motifs from protein coordinate data: Secondary structure and first-level supersecondary structure. Proteins Struct. Funct. Bioinform. 1988, 3, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Labesse, G.; Colloc’h, N.; Pothier, J.; Mornon, J.P. P-SEA: A new efficient assignment of secondary structure from Cα trace of proteins. Bioinformatics 1997, 13, 291–295. [Google Scholar] [CrossRef]
- Martin, J.; Letellier, G.; Marin, A.; Taly, J.F.; de Brevern, A.G.; Gibrat, J.F. Protein secondary structure assignment revisited: A detailed analysis of different assignment methods. BMC Struct. Biol. 2005, 5, 17. [Google Scholar] [CrossRef]
- Cao, C.; Wang, G.; Liu, A.; Xu, S.; Wang, L.; Zou, S. A New Secondary Structure Assignment Algorithm Using Cα Backbone Fragments. Int. J. Mol. Sci. 2016, 17, 333. [Google Scholar] [CrossRef]
- Taylor, W.R. Defining linear segments in protein structure. J. Mol. Biol. 2001, 310, 1135–1150. [Google Scholar] [CrossRef]
- Konagurthu, A.S.; Allison, L.; Stuckey, P.J.; Lesk, A.M. Piecewise linear approximation of protein structures using the principle of minimum message length. Bioinformatics 2011, 27, i43–i51. [Google Scholar] [CrossRef]
- Si, D.; Ji, S.; Al Nasr, K.; He, J. A machine learning approach for the identification of protein secondary structure elements from cryoEM density maps. Biopolymers 2012, 97, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Saqib, M.N.; Kryś, J.D.; Gront, D. Automated Protein Secondary Structure Assignment from Cα Positions Using Neural Networks. Biomolecules 2022, 12, 841. [Google Scholar] [CrossRef] [PubMed]
- Salawu, E.O. RaFoSA: Random forests secondary structure assignment for coarse-grained and all-atom protein systems. Cogent Biol. 2016, 2, 1214061. [Google Scholar] [CrossRef]
- Sallal, M.A.; Chen, W.; Al Nasr, K. Machine Learning Approach to Assign Protein Secondary Structure Elements from Cα Trace. In Proceedings of the 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Seoul, Republic of Korea, 16–19 December 2020; pp. 35–41. [Google Scholar]
- Sekmen, A.; Al Nasr, K.; Jones, C. Subspace Modeling for Classification of Protein Secondary Structure Elements from Cα Trace. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021. [Google Scholar]
- Al Nasr, K.; Sekmen, A.; Bilgin, B.; Jones, C.; Koku, A.B. Deep Learning for Assignment of Protein Secondary Structure Elements from Cα Coordinates. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021. [Google Scholar]
- Vidal, R.; Ma, Y.; Sastry, S. Generalized Principal Component Analysis (GPCA). IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1945–1959. [Google Scholar] [CrossRef] [PubMed]
- Roy, O.; Vetterli, M. The effective rank: A measure of effective dimensionality. In Proceedings of the 2007 15th European Signal Processing Conference, Poznan, Poland, 3–7 September 2007; pp. 606–610. [Google Scholar]
- Berner, J.; Grohs, P.; Kutyniok, G.; Petersen, P. The modern mathematics of deep learning. arXiv 2021, arXiv:2105.04026. [Google Scholar]
- Ho, J.; Yang, M.; Lim, J.; Kriegman, D. Clustering appearances of objects under varying illumination conditions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Madison, WI, USA, 18–20 June 2003; pp. 11–18. [Google Scholar]
- Aldroubi, A.; Sekmen, A. Nearness to local subspace algorithm for subspace and motion segmentation. IEEE Signal Process. Lett. 2012, 19, 704–707. [Google Scholar] [CrossRef]
- Vidal, R. A tutorial on subspace clustering. IEEE Signal Process. Mag. 2010, 28, 52–68. [Google Scholar] [CrossRef]
- Georghiades, A.S.; Belhumeur, P.N.; Kriegman, D.J. From Few to Many: Illumination Cone Models for Face Recognition under Variable Lighting and Pose. IEEE Trans. Pattern Anal. Mach. Intell. 2001, 23, 643–660. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, G.; Heath, R.W., Jr.; Huang, K. Grassmannian Learning: Embedding Geometry Awareness in Shallow and Deep Learning. arXiv 2018, arXiv:1808.02229. [Google Scholar]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef]
- Bolstad, B.; Irizarry, R.; Åstrand, M.; Speed, T. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, D.H. Stacked generalization. Neural Netw. 1992, 5, 241–259. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Network ID | Validation Accuracy |
|---|---|
| #1 | |
| #2 | |
| #3 | |
| #4 | |
| #5 | |
| #6 | |
| #7 | |
| #8 | |
| #9 | |
| #10 | |
| Joint Prediction |
| Precision | Recall | F1-Score | Accuracy | |
|---|---|---|---|---|
| Helix | ||||
| Sheet | ||||
| Loop |
| Observed/Predicted | Helix | Sheet | Loop | Total |
|---|---|---|---|---|
| Helix | 26,681 (84.65%) | 286 (0.90%) | 4554 (14.45%) | 31,521 |
| Sheet | 8 (0.06%) | 12,022 (83.64%) | 2343 (16.30%) | 14,373 |
| Loop | 1001 (4.41%) | 2868 (12.65%) | 18,809 (82.94%) | 22,678 |
| Total | 27,690 | 15,176 | 25,706 | 68,572 |
| Precision | Recall | F1-Score | Accuracy | |
|---|---|---|---|---|
| Helix | ||||
| Sheet | ||||
| Loop |
| Observed/Predicted | Helix | Sheet | Loop | Total |
|---|---|---|---|---|
| Helix | 26,082 (82.74%) | 248 (0.79%) | 5191 (16.47%) | 31,521 |
| Sheet | 26 (0.18%) | 11,043 (76.83%) | 3304 (22.99%) | 14,373 |
| Loop | 1210 (5.34%) | 4665 (20.57%) | 16,803 (74.09%) | 22,678 |
| Total | 27,318 | 15,956 | 25,298 | 68,572 |
| Precision | Recall | F1-Score | Accuracy | |
|---|---|---|---|---|
| Helix | ||||
| Sheet | ||||
| Loop |
| Observed/Predicted | Helix | Sheet | Loop | Total |
|---|---|---|---|---|
| Helix | 30,918 (98.09%) | 24 (0.07%) | 579 (1.84%) | 31,521 |
| Sheet | 44 (0.31%) | 13,274 (92.35%) | 1055 (7.34%) | 14,373 |
| Loop | 804 (3.55%) | 842 (3.71%) | 21,032 (92.74%) | 22,678 |
| Total | 31,766 | 14,140 | 22,666 | 68,572 |
| Precision | Recall | F1-Score | Accuracy | |
|---|---|---|---|---|
| Helix | ||||
| Sheet | ||||
| Loop |
| Observed/Predicted | Helix | Sheet | Loop | Total |
|---|---|---|---|---|
| Helix | 38,889 (97.22%) | 24 (0.06%) | 1087 (2.72%) | 40,000 |
| Sheet | 17 (0.04%) | 38,895 (97.24%) | 1088 (2.72%) | 40,000 |
| Loop | 1070 (2.67%) | 1129 (2.82%) | 37,801 (94.50%) | 40,000 |
| Total | 39,976 | 40,048 | 39,976 | 120,000 |
| Precision | Recall | F1-Score | Accuracy | |
|---|---|---|---|---|
| Helix | ||||
| Sheet | ||||
| Loop |
| Observed/Predicted | Helix | Sheet | Loop | Total |
|---|---|---|---|---|
| Helix | 29,654 (95.85%) | 26 (0.08%) | 1259 (4.07%) | 30,939 |
| Sheet | 15 (0.11%) | 13,064 (92.93%) | 979 (6.96%) | 14,058 |
| Loop | 935 (4.45%) | 1066 (5.08%) | 18,998 (90.47%) | 20,999 |
| Total | 30,604 | 14,156 | 21,236 | 65,996 |
| Precision | Recall | F1-Score | Accuracy | |
|---|---|---|---|---|
| Helix | ||||
| Sheet | ||||
| Loop |
| Observed/Predicted | Helix | Sheet | Loop | Total |
|---|---|---|---|---|
| Helix | 24,702 (77.67%) | 349 (1.10%) | 6751 (21.23%) | 31,802 |
| Sheet | 12 (0.09%) | 12,233 (86.35%) | 1921 (13.56%) | 14,166 |
| Loop | 413 (1.92%) | 1145 (5.31%) | 20,001 (92.77%) | 21,559 |
| Total | 25,127 | 14,036 | 28,673 | 67,526 |
| Dataset | EML | Model-1 | Model-2 | DL | PCASSO |
|---|---|---|---|---|---|
| S | 93.51 | 83.9 | 78.6 | 95.12 | 84.3 |
| Num | Protein ID a | Chain ID b | #AA c | EML% d | Subspace I% e | Subspace II% f | DL% g | PCASSO% h |
|---|---|---|---|---|---|---|---|---|
| 1 | 3SSB | I | 30 | 75.0 | 83.9 | 78.6 | 80.0 | 26.7 |
| 2 | 2END | A | 137 | 75.6 | 83.8 | 78.7 | 76.6 | 81.8 |
| 3 | 1ZUU | A | 56 | 80.0 | 84.0 | 78.8 | 78.6 | 80.4 |
| 4 | 4UE8 | B | 37 | 63.3 | 83.7 | 78.5 | 67.6 | 48.6 |
| 5 | 5W82 | A | 100 | 77.4 | 84.0 | 78.8 | 77.0 | 94.0 |
| 6 | 3QR7 | A | 115 | 80.7 | 84.1 | 78.8 | 73.9 | 95.2 |
| 7 | 3NGG | A | 46 | 82.5 | 83.9 | 78.8 | 87.0 | 84.4 |
| 8 | 3X34 | A | 87 | 81.3 | 84.1 | 78.8 | 83.9 | 63.3 |
| 9 | 1KVE | A | 63 | 75.5 | 83.9 | 78.7 | 73.0 | 90.5 |
| 10 | 5DBL | A | 130 | 86.3 | 84.0 | 78.7 | 94.6 | 66.7 |
| 11 | 4KK7 | A | 385 | 93.7 | 83.7 | 78.5 | 94.0 | 88.8 |
| 12 | 3MAO | A | 105 | 91.9 | 84.0 | 78.8 | 95.2 | 87.6 |
| 13 | 5QS9 | A | 171 | 95.2 | 83.8 | 78.7 | 96.0 | 90.6 |
| 14 | 6DWD | D | 481 | 94.2 | 83.5 | 78.4 | 96.7 | 85.2 |
| 15 | 4G9S | B | 111 | 98.1 | 83.6 | 78.7 | 98.2 | 30.1 |
| 16 | 3ZVS | A | 158 | 94.7 | 83.9 | 78.7 | 96.2 | 96.6 |
| 17 | 3QL9 | A | 125 | 93.3 | 84.0 | 78.7 | 93.6 | 87.9 |
| 18 | 4ZFL | A | 229 | 94.1 | 83.8 | 78.7 | 93.4 | 86.7 |
| 19 | 5OBY | A | 365 | 94.1 | 83.5 | 78.5 | 96.7 | 86.1 |
| 20 | 6B1K | A | 114 | 93.3 | 84.2 | 78.8 | 90.4 | 85.1 |
| 21 | 4ONR | A | 147 | 100 | 84.0 | 78.8 | 99.3 | 93.8 |
| 22 | 2IC6 | A | 71 | 100 | 84.1 | 78.8 | 100 | 94.4 |
| 23 | 3HE5 | B | 48 | 100 | 84.2 | 78.9 | 97.9 | 95.7 |
| 24 | 3LDC | A | 82 | 100 | 84.0 | 78.8 | 100 | 91.5 |
| 25 | 4ABM | A | 79 | 100 | 83.8 | 78.8 | 100 | 94.9 |
| 26 | 4I6R | A | 77 | 100 | 84.0 | 78.7 | 100 | 62.0 |
| 27 | 4WZX | A | 87 | 100 | 84.0 | 78.8 | 98.9 | 87.2 |
| 28 | 5OI7 | A | 88 | 100 | 83.9 | 78.5 | 98.9 | 95.4 |
| 29 | 3D3B | A | 139 | 100 | 84.2 | 78.8 | 100 | 84.2 |
| 30 | 5XAU | B | 71 | 100 | 84.1 | 78.8 | 98.6 | 97.2 |
| Average | 131.13 | 90.7 | 83.9 | 78.7 | 91.2 | 81.8 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekmen, A.; Al Nasr, K.; Bilgin, B.; Koku, A.B.; Jones, C. Mathematical and Machine Learning Approaches for Classification of Protein Secondary Structure Elements from Cα Coordinates. Biomolecules 2023, 13, 923. https://doi.org/10.3390/biom13060923
Sekmen A, Al Nasr K, Bilgin B, Koku AB, Jones C. Mathematical and Machine Learning Approaches for Classification of Protein Secondary Structure Elements from Cα Coordinates. Biomolecules. 2023; 13(6):923. https://doi.org/10.3390/biom13060923
Chicago/Turabian StyleSekmen, Ali, Kamal Al Nasr, Bahadir Bilgin, Ahmet Bugra Koku, and Christopher Jones. 2023. "Mathematical and Machine Learning Approaches for Classification of Protein Secondary Structure Elements from Cα Coordinates" Biomolecules 13, no. 6: 923. https://doi.org/10.3390/biom13060923
APA StyleSekmen, A., Al Nasr, K., Bilgin, B., Koku, A. B., & Jones, C. (2023). Mathematical and Machine Learning Approaches for Classification of Protein Secondary Structure Elements from Cα Coordinates. Biomolecules, 13(6), 923. https://doi.org/10.3390/biom13060923