
Epigenetic Reader Bromodomain-Containing Protein 4 in Aging-Related Vascular Pathologies and Diseases: Molecular Basis, Functional Relevance, and Clinical Potential


Abstract
:1. Introduction
2. Molecular Basis for the Biological Functions of BRD4
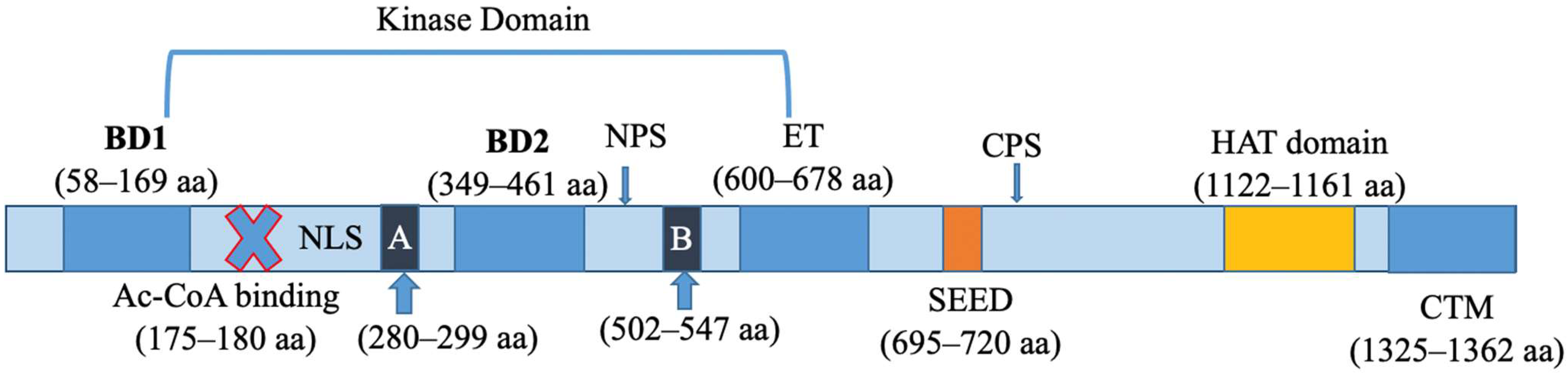
2.1. Molecular Structure
2.2. Biological Function of BRD4 as an Epigenomic Reader
2.2.1. Histone Acetylation
2.2.2. Gene Transcription
2.2.3. Alternative Splicing
2.3. Functional Relevance of the Modifications in BRD4 Molecule
2.3.1. Post-Translational Modifications (PTMs) of BRD4
2.3.2. Alternative Splicing of BRD4
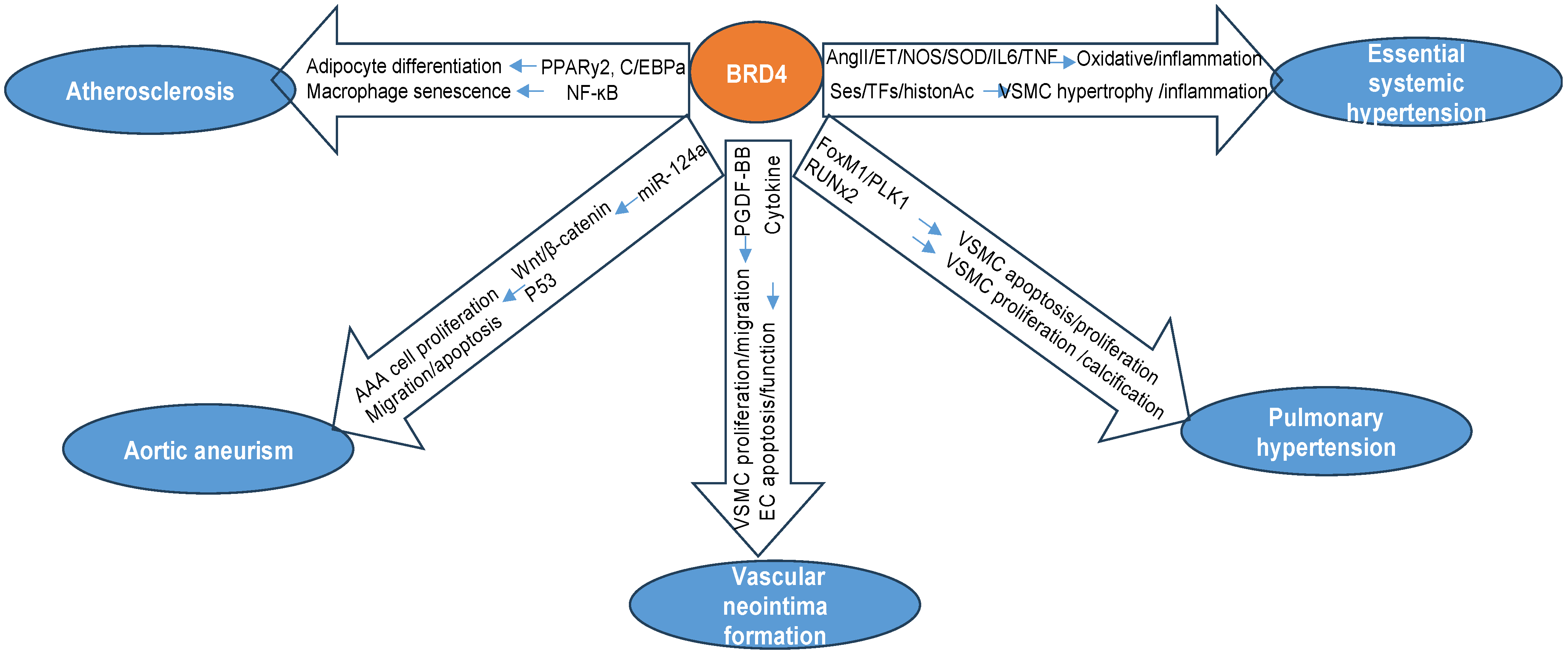
3. BRD4 in Aging-Related Vascular Pathologies and Diseases
3.1. Atherosclerosis
3.2. Aortic Aneurism
3.3. Vascular Neointima Formation
3.4. Pulmonary Arterial Hypertension (PAH)
3.5. Essential/Systemic Hypertension
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| AAA | Abdominal aortic aneurysm |
| ACBP | Acetyl-CoA binding protein |
| AML | Acute myeloid leukemia |
| AP1 | Activator protein 1 |
| BAT | Brown adipose tissue |
| BD | Bromo domain |
| BET | Bromodomain and Extra-Terminal domain |
| BID | Bromo-interacting domain |
| BMI | Body mass index |
| BMPR2 BRD4L BRD4S | Bone morphogenic protein receptor 2 Bromo-domain protein 4 long isoform bromo-domain protein 4 short isoform |
| BRDT BRD2 BRD3 BRD4 CDX2 | Bromodomain testis-specific protein Bromodomain-containing protein 2 Bromodomain-containing protein 3 Bromodomain-containing protein 4 Caudal-type homeobox 2 |
| C/EBP-α | CCATT-enhancer-binding protein alpha |
| CHD4 | Chromodomain-helicase-DNA-binding protein 4 |
| CK2 | Casein kinase 2 |
| CPS | C-terminal cluster of phosphorylation sites |
| CTD | Carboxy-terminal domain |
| CTM | C-terminal motif |
| DC | Dyskeratosis congenita |
| DCM | Dilated cardiomyopathy |
| DDR | DNA damage response |
| EC | Endothelial cells |
| EH EGFR | Essential hypertension Epidermal growth factor receptor |
| EMT EN1 | Epithelial-mesenchymal transitions Engrailed homeobox 1 |
| ESC | Embryonic stem cells |
| ET | Extra-terminal |
| ETS FOS FOXM1 | Erythroblast transformation specific FBJ murine osteosarcoma viral oncogene homolog Forkhead box M1 |
| FUS | Fused in sarcoma |
| GLTSCR1 | Glioma tumor suppressor candidate region gene 1 |
| H3HATs | Histone 3 acetyltransferases |
| H3K14 H3K122 H3K27ac H3K27M | Histone 3 lysine 14 acetylation Histone 3 lysine 122 acetylation Histone 3 lysine 27 acetylation Histone 3 lysine 27 methionine |
| H3K4me1 HF | Histone 3 lysine 4 monomethylation Heart Failure |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reserved ejection fraction |
| HIV-1 | Human immunodeficiency virus 1 |
| HnRNPM HPV I-BET762 IDH | Heterogenous nuclear ribonucleoprotein M Human papillomavirus Selective inhibitor of bromodomain extra-terminal domain 762 Isocitrate dehydrogenase |
| IH | Intimal hyperplasia |
| IPAH ISX | Idiopathic pulmonary arterial hypertension Intestine-specific homeobox |
| JQ-1 | Jun Qi 1 |
| KO | Knockout |
| LLPS | Liquid–liquid phase separations |
| LPS | Lipopolysaccharide |
| MDA | Malondialdehyde |
| MI MYC | Myocardial infraction Myelocytomatosis oncogene |
| miRNA | Micro RNA |
| MVECs | Microvascular endothelial cells |
| NF-kB | Nuclear factor- kappa B |
| NLS | Nuclear localization signal |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NPS | N-terminal cluster of phosphorylation sites |
| PAH | Pulmonary arterial hypertension |
| PBMCs | Peripheral blood mononuclear cells |
| PCAF | P300/CBP-associated factor |
| PDGF-BB PDID | Platelet derived growth factor Phosphor-dependent interaction domain |
| PLA | Proximity ligation assays |
| PLK1 | Polo-like kinase 1 |
| PPARy2 | Peroxisome proliferator activated receptor gamma |
| Prdm14 | PR domain zinc finger protein 14 |
| P-TEFb | Positive transcription elongation factor |
| PTMs | Post-translational modifications |
| PVR | Pulmonary vascular resistance |
| RUNX2 | Runt-related transcription factor 2 |
| RVF | Right ventricle failure |
| SASP | Senescence-associated secretory phenotype |
| SE | Super enhancer |
| SEED | Ser (S)/Glu (E)/Asp (D) |
| SH-PAH | Sugen hypoxia-pulmonary arterial hypertension |
| SHRs | Spontaneously hypertensive rats |
| SMCs | Smooth muscle cells |
| SOD | Superoxide dismutase |
| SPOP-DUB3 | Speckle-type BTB/POZ protein-de-ubiquitination enzyme 3 |
| STAT1 | Signal transducer and activator of transcription 1 |
| TAC | Transverse aortic constriction |
| TALL | T cell acute lymphoblastic leukemia |
| TF | Transcription factor |
| VSMC Wnt/β-catenin | Vascular smooth muscle cell Wingless-related integration site/ beta- catenin |
| YY1 | Ying-yang 1 |
References
- Qiu, H.; Depre, C.; Ghosh, K.; Resuello, R.G.; Natividad, F.F.; Rossi, F.; Peppas, A.; Shen, Y.T.; Vatner, D.E.; Vatner, S.F. Mechanism of gender-specific differences in aortic stiffness with aging in nonhuman primates. Circulation 2007, 116, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Rich, M.W.; Chyun, D.A.; Skolnick, A.H.; Alexander, K.P.; Forman, D.E.; Kitzman, D.W.; Maurer, M.S.; McClurken, J.B.; Resnick, B.M.; Shen, W.K.; et al. Knowledge Gaps in Cardiovascular Care of the Older Adult Population: A Scientific Statement From the American Heart Association, American College of Cardiology, and American Geriatrics Society. J. Am. Coll. Cardiol. 2016, 67, 2419–2440. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Chen, J.; Li, B.Y.; Wood, S.C.; Rosebury-Smith, W.; Remmer, H.A.; Jiang, L.; Zhang, M.; Salmon, M.; Ailawadi, G.; et al. Aging Alters the Aortic Proteome in Health and Thoracic Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1060–1076. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Melton, E.; Wiener, R.; Zhou, N.; Wu, W.; Lai, L.; Wang, C.; Costa, K.D.; Qiu, H. Age and Blood Pressure Contribute to Aortic Cell and Tissue Stiffness Through Distinct Mechanisms. Hypertension 2022, 79, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Melton, E.; Qiu, H. Interleukin-1beta in Multifactorial Hypertension: Inflammation, Vascular Smooth Muscle Cell and Extracellular Matrix Remodeling, and Non-Coding RNA Regulation. Int. J. Mol. Sci. 2021, 22, 8639. [Google Scholar] [CrossRef]
- Onuh, J.O.; Qiu, H. New progress on the study of aortic stiffness in age-related hypertension. J. Hypertens. 2020, 38, 1871–1877. [Google Scholar] [CrossRef]
- Qiu, H.; Tian, B.; Resuello, R.G.; Natividad, F.F.; Peppas, A.; Shen, Y.T.; Vatner, D.E.; Vatner, S.F.; Depre, C. Sex-specific regulation of gene expression in the aging monkey aorta. Physiol. Genom. 2007, 29, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Stoll, S.; Wang, C.; Qiu, H. DNA Methylation and Histone Modification in Hypertension. Int. J. Mol. Sci. 2018, 19, 1174. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Qiu, H.; Trzeciakowski, J.P.; Sun, Z.; Li, Z.; Hong, Z.; Hill, M.A.; Hunter, W.C.; Vatner, D.E.; Vatner, S.F.; et al. Temporal analysis of vascular smooth muscle cell elasticity and adhesion reveals oscillation waveforms that differ with aging. Aging Cell 2012, 11, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Faggion Vinholo, T.; Brownstein, A.J.; Ziganshin, B.A.; Zafar, M.A.; Kuivaniemi, H.; Body, S.C.; Bale, A.E.; Elefteriades, J.A. Genes Associated with Thoracic Aortic Aneurysm and Dissection: 2019 Update and Clinical Implications. Aorta 2019, 7, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.M.D.; Chen, Y.C.; Peter, K. Vascular Aging and Vascular Disease Have Much in Common! Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1077–1080. [Google Scholar] [CrossRef]
- Thorpe, K.L.; Abdulla, S.; Kaufman, J.; Trowsdale, J.; Beck, S. Phylogeny and structure of the RING3 gene. Immunogenetics 1996, 44, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Salazar, G.; Crowley, T.E.; Wang, X.; Lopez, R.A.; Wang, X.; Wolgemuth, D.J. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr. Patterns 2004, 4, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.H.; Numata, M.; Shimane, M. Identification and characterization of BRDT: A testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics 1997, 45, 529–534. [Google Scholar] [CrossRef]
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [Green Version]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaiah, B.N.; Gegonne, A.; Singer, D.S. Bromodomain 4: A cellular Swiss army knife. J. Leukoc. Biol. 2016, 100, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Park, Y.K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 2217. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Etxaniz, U.; Dall’Agnese, A.; Wu, S.Y.; Chiang, C.M.; Brennan, P.E.; Wood, M.J.A.; Puri, P.L. BRD3 and BRD4 BET Bromodomain Proteins Differentially Regulate Skeletal Myogenesis. Sci. Rep. 2017, 7, 6153. [Google Scholar] [CrossRef]
- Lu, L.; Chen, Z.; Lin, X.; Tian, L.; Su, Q.; An, P.; Li, W.; Wu, Y.; Du, J.; Shan, H.; et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020, 27, 255–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drumond-Bock, A.L.; Bieniasz, M. The role of distinct BRD4 isoforms and their contribution to high-grade serous ovarian carcinoma pathogenesis. Mol. Cancer 2021, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Situ, Y.; Liang, Q.; Zeng, Z.; Chen, J.; Shao, Z.; Xu, Q.; Lu, X.; Cui, Y.; Zhang, J.; Lu, L.; et al. Systematic analysis of the BET family in adrenocortical carcinoma: The expression, prognosis, gene regulation network, and regulation targets. Front. Endocrinol. 2023, 14, 1089531. [Google Scholar] [CrossRef]
- Yu, X.; Long, Q.; Shen, S.; Liu, Z.; Chandran, J.; Zhang, J.; Ding, H.; Zhang, H.; Cai, D.; Kim, E.S.; et al. Screening of an epigenetic compound library identifies BRD4 as a potential antiviral target for hepatitis B virus covalently closed circular DNA transcription. Antiviral. Res. 2023, 211, 105552. [Google Scholar] [CrossRef]
- Meta-analysis Global Group in Chronic Heart Failure. The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: An individual patient data meta-analysis. Eur. Heart J. 2012, 33, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Du, L. The therapeutic potential of BRD4 in cardiovascular disease. Hypertens. Res. 2020, 43, 1006–1014. [Google Scholar] [CrossRef]
- Ijaz, T.; Burke, M.A. BET Protein-Mediated Transcriptional Regulation in Heart Failure. Int. J. Mol. Sci. 2021, 22, 6059. [Google Scholar] [CrossRef]
- Li, L.; Xie, W.; Gui, Y.; Zheng, X.L. Bromodomain-containing protein 4 and its role in cardiovascular diseases. J. Cell. Physiol. 2021, 236, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Josling, G.A.; Selvarajah, S.A.; Petter, M.; Duffy, M.F. The role of bromodomain proteins in regulating gene expression. Genes 2012, 3, 320–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Umehara, T.; Nakano, K.; Jang, M.K.; Shirouzu, M.; Morita, S.; Uda-Tochio, H.; Hamana, H.; Terada, T.; Adachi, N.; et al. Crystal structure of the human BRD2 bromodomain: Insights into dimerization and recognition of acetylated histone H4. J. Biol. Chem. 2007, 282, 4193–4201. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Konuma, T.; Yu, D.; Zhao, C.; Ju, Y.; Sharma, R.; Ren, C.; Zhang, Q.; Zhou, M.M.; Zeng, L. Structural Mechanism of the Oxygenase JMJD6 Recognition by the Extraterminal (ET) Domain of BRD4. Sci. Rep. 2017, 7, 16272. [Google Scholar] [CrossRef] [Green Version]
- Luna-Pelaez, N.; March-Diaz, R.; Ceballos-Chavez, M.; Guerrero-Martinez, J.A.; Grazioli, P.; Garcia-Gutierrez, P.; Vaccari, T.; Massa, V.; Reyes, J.C.; Garcia-Dominguez, M. The Cornelia de Lange Syndrome-associated factor NIPBL interacts with BRD4 ET domain for transcription control of a common set of genes. Cell Death Dis. 2019, 10, 548. [Google Scholar] [CrossRef] [Green Version]
- Crowe, B.L.; Larue, R.C.; Yuan, C.; Hess, S.; Kvaratskhelia, M.; Foster, M.P. Structure of the Brd4 ET domain bound to a C-terminal motif from gamma-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. USA 2016, 113, 2086–2091. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [Green Version]
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021, 41, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppal, S.; Gegonne, A.; Chen, Q.; Thompson, P.S.; Cheng, D.; Mu, J.; Meerzaman, D.; Misra, H.S.; Singer, D.S. The Bromodomain Protein 4 Contributes to the Regulation of Alternative Splicing. Cell Rep. 2019, 29, 2450–2460.e2455. [Google Scholar] [CrossRef] [Green Version]
- Malvezzi, F.; Stubbs, C.J.; Jowitt, T.A.; Dale, I.L.; Guo, X.; DeGnore, J.P.; Degliesposti, G.; Skehel, J.M.; Bannister, A.J.; McAlister, M.S. Phosphorylation-dependent BRD4 dimerization and implications for therapeutic inhibition of BET family proteins. Commun. Biol. 2021, 4, 1273. [Google Scholar] [CrossRef]
- Wang, W.; Tang, Y.A.; Xiao, Q.; Lee, W.C.; Cheng, B.; Niu, Z.; Oguz, G.; Feng, M.; Lee, P.L.; Li, B.; et al. Stromal induction of BRD4 phosphorylation Results in Chromatin Remodeling and BET inhibitor Resistance in Colorectal Cancer. Nat. Commun. 2021, 12, 4441. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Nikonova, A.S.; Zhang, P.; Deneka, A.Y.; Fitzgerald, M.E.; Michael, R.E.; Lee, L.; Lilly, A.C.; Fisher, S.L.; Phillips, A.J.; et al. Evaluation of the Small-molecule BRD4 Degrader CFT-2718 in Small-cell Lung Cancer and Pancreatic Cancer Models. Mol. Cancer Ther. 2021, 20, 1367–1377. [Google Scholar] [CrossRef]
- Sanz-Alvarez, M.; Cristobal, I.; Luque, M.; Santos, A.; Zazo, S.; Madoz-Gurpide, J.; Carames, C.; Chiang, C.M.; Garcia-Foncillas, J.; Eroles, P.; et al. Expression of Phosphorylated BRD4 Is Markedly Associated with the Activation Status of the PP2A Pathway and Shows a Strong Prognostic Value in Triple Negative Breast Cancer Patients. Cancers 2021, 13, 1246. [Google Scholar] [CrossRef]
- Malik, R.; Kopylov, M.; Gomez-Llorente, Y.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Structure and mechanism of B-family DNA polymerase zeta specialized for translesion DNA synthesis. Nat. Struct. Mol. Biol. 2020, 27, 913–924. [Google Scholar] [CrossRef]
- Liu, N.; Ling, R.; Tang, X.; Yu, Y.; Zhou, Y.; Chen, D. Post-Translational Modifications of BRD4: Therapeutic Targets for Tumor. Front. Oncol. 2022, 12, 847701. [Google Scholar] [CrossRef]
- Tzelepis, K.; De Braekeleer, E.; Aspris, D.; Barbieri, I.; Vijayabaskar, M.S.; Liu, W.H.; Gozdecka, M.; Metzakopian, E.; Toop, H.D.; Dudek, M.; et al. SRPK1 maintains acute myeloid leukemia through effects on isoform usage of epigenetic regulators including BRD4. Nat. Commun. 2018, 9, 5378. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Fu, X.; Yang, R.; Zhang, W. Atherosclerosis Vascular Endothelial Secretion Dysfunction and Smooth Muscle Cell Proliferation. J. Healthc. Eng. 2022, 2022, 9271879. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Feldman, Z.B.; Doherty, S.P.; Reyes, J.M.; Rahl, P.B.; Lin, C.Y.; Sheng, Q.; Duan, Q.; Federation, A.J.; Kung, A.L.; et al. BET bromodomain proteins regulate enhancer function during adipogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 2144–2149. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Fu, H.; Zhu, R.; Wu, X.; Ji, X.; Li, X.; Jiang, H.; Lin, Z.; Tang, X.; Sun, S.; et al. BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 2020, 12, 9240–9259. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lv, M.; Lu, M.; Guan, H. miR-124a Involves in the Regulation of Wnt/beta-Catenin and P53 Pathways to Inhibit Abdominal Aortic Aneurysm via Targeting BRD4. Comput. Math. Methods Med. 2022, 2022, 9241959. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Moehle, C.W.; Johnson, J.L.; Yang, Z.; Lee, J.K.; Jackson, C.L.; Owens, G.K. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Investig. 2012, 122, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Zhang, M.; Takayama, T.; Shi, X.; Roenneburg, D.A.; Kent, K.C.; Guo, L.W. BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine 2015, 2, 1650–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Wang, B.; Urabe, G.; Huang, Y.; Plutzky, J.; Kent, K.C.; Guo, L.W. The BD2 domain of BRD4 is a determinant in EndoMT and vein graft neointima formation. Cell Signal. 2019, 61, 20–29. [Google Scholar] [CrossRef]
- Verma, S.; Lovren, F.; Pan, Y.; Yanagawa, B.; Deb, S.; Karkhanis, R.; Quan, A.; Teoh, H.; Feder-Elituv, R.; Moussa, F.; et al. Pedicled no-touch saphenous vein graft harvest limits vascular smooth muscle cell activation: The PATENT saphenous vein graft study. Eur. J. Cardiothorac. Surg. 2014, 45, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Zhao, Y.Y. BET in Pulmonary Arterial Hypertension: Exploration of BET Inhibitors to Reverse Vascular Remodeling. Am. J. Respir. Crit. Care Med. 2019, 200, 806–808. [Google Scholar] [CrossRef]
- Meloche, J.; Potus, F.; Vaillancourt, M.; Bourgeois, A.; Johnson, I.; Deschamps, L.; Chabot, S.; Ruffenach, G.; Henry, S.; Breuils-Bonnet, S.; et al. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ. Res. 2015, 117, 525–535. [Google Scholar] [CrossRef]
- Stratton, M.S.; McKinsey, T.A. Acetyl-lysine erasers and readers in the control of pulmonary hypertension and right ventricular hypertrophy. Biochem. Cell Biol. 2015, 93, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L.; Szulcek, R.; Bourgeois, A.; Lampron, M.C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Ruffenach, G.; Chabot, S.; Tanguay, V.F.; Courboulin, A.; Boucherat, O.; Potus, F.; Meloche, J.; Pflieger, A.; Breuils-Bonnet, S.; Nadeau, V.; et al. Role for Runt-related Transcription Factor 2 in Proliferative and Calcified Vascular Lesions in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1273–1285. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, J.Y.; Jeon, H.J.; Cho, B.M.; Park, S.H.; Oh, S.M. Intraoperative Endovascular Embolization of Middle Meningeal Artery and a Pseudoaneurysm by Using N-Butyl 2-Cyanoacrylate for Hemostasis during Operation of Acute Epidural Hemorrhage. Korean J. Neurotrauma 2015, 11, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Sancisi, V.; Manzotti, G.; Gugnoni, M.; Rossi, T.; Gandolfi, G.; Gobbi, G.; Torricelli, F.; Catellani, F.; Faria do Valle, I.; Remondini, D.; et al. RUNX2 expression in thyroid and breast cancer requires the cooperation of three non-redundant enhancers under the control of BRD4 and c-JUN. Nucleic Acids Res. 2017, 45, 11249–11267. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, X.; Liu, Z.; Huang, L.; Yao, Y.; Li, L.; Chen, J.; Zhang, R.; Zhou, J.; Wang, L.; et al. A Bromodomain-Containing Protein 4 (BRD4) Inhibitor Suppresses Angiogenesis by Regulating AP-1 Expression. Front. Pharmacol. 2020, 11, 1043. [Google Scholar] [CrossRef]
- Yang, Y.M.; Shi, R.H.; Xu, C.X.; Li, L. BRD4 expression in patients with essential hypertension and its effect on blood pressure in spontaneously hypertensive rats. J. Am. Soc. Hypertens. 2018, 12, e107–e117. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Das, S.; Senapati, P.; Chen, Z.; Reddy, M.A.; Ganguly, R.; Lanting, L.; Mandi, V.; Bansal, A.; Leung, A.; Zhang, S.; et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Suarez-Alvarez, B.; Morgado-Pascual, J.L.; Rayego-Mateos, S.; Rodriguez, R.M.; Rodrigues-Diez, R.; Cannata-Ortiz, P.; Sanz, A.B.; Egido, J.; Tharaux, P.L.; Ortiz, A.; et al. Inhibition of Bromodomain and Extraterminal Domain Family Proteins Ameliorates Experimental Renal Damage. J. Am. Soc. Nephrol. 2017, 28, 504–519. [Google Scholar] [CrossRef]
- Duan, Q.; Mao, X.; Liao, C.; Zhou, H.; Sun, Z.; Deng, X.; Hu, Q.; Qi, J.; Zhang, G.; Huang, H.; et al. Inhibition of BET bromodomain attenuates angiotensin II induced abdominal aortic aneurysm in ApoE(-/-) mice. Int. J. Cardiol. 2016, 223, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Lyv, X.; Zhou, Q.J.; Xiang, Z.; Stanford, D.; Bodduluri, S.; Rowe, S.M.; Thannickal, V.J. Brd4-p300 inhibition downregulates Nox4 and accelerates lung fibrosis resolution in aged mice. JCI Insight 2020, 5, e137127. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Scaffidi, P.; Markert, E.; Lee, J.H.; Rane, S.; Misteli, T. Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4. Cell Rep. 2014, 9, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| S/N | RESULTS | REFERENCES | |
|---|---|---|---|
| 1 | Ang II Infusion | A. Increased BRD4 are reported by several groups in SHR rats and mice treated with AngII. B. AngII stimulation increased the recruitment of transcription factors at their respective binding sites. BRD4 is recruited, enhancing the transcription of nearby genes in VSMCs. C. Early inflammation was produced by AngII; by 3 days, more infiltrating interstitial monocytes/ macrophages and proinflammatory mediators were seen than in mice treated with JQ-1. | 1. [67] Yang et al. 2018 2. [69,70,71]; Brown et al. 2014 Loven et al. 2013 Das et al. 2017 3. [72] Suarez-Alvarez et al. 2017 |
| 2 | Carotid Artery Angioplasty | A. BRD4 was significantly increased in neointima formation, and blocking BRD4 with JQ-1 inhibited IH, by diminishing proliferation and migration of VSMCs. JQ-1 treatment induced a pronounced inhibitory effect on the proliferation and migration of rat and human primary aortic VSMCs. | 1. [56] Wang et al. 2015 |
| 3 | Adipogenesis /Myogenesis cell Differentiation | A. BRD4 functions as an enhancer epigenomic reader that links active enhancers to the activation of the cell identity gene during differentiation. BRD4 is required for BAT and muscle development, as evidenced muscle mass were severely reduced when BRD4 is deleted in the cervical regions of E18.5 embryos. B. RNA interference-based specific BET protein knockdown demonstrated that BRD4 was necessary for myogenic differentiation. | 1. [23] Lee et al. 2017 2. [24] Roberts et al. 2017 |
| 4 | EndoMT Vein Graft | A. BRD4 control of mediators and indicators of EndoMT, showed that BRD4 modification might diminish neointima development in vein grafts in vivo given recent evidence that EndoMT is essential for neointima formation. B. MicroRNA -145 encouraged the binding of myocardin to SRF. Additionally, it suppressed KLF4, which through blocking myocardin and SRF, promoted aberrant VSMC development. | 1. [57] Zhang et al. 2019 2. [58] Verma et al. 2013 |
| 5 | Infection-Induced Senescent Macrophages | A. By increasing BRD4 expression, the models used for LPS treatment showed to induce cellular senescence by encouraging the formation of SASP and the advancement of atherosclerosis-like conditions. | 1. [53] Wang et al. 2020 |
| 6 | Abdominal Aortic Aneurysm (AAA) cells | A. Downregulation of miR-124a was present in whole blood of patients and decreased in AAA cell models. The confirmation of the dual-luciferase reporter assay depicted that BRD4 was a downstream target of miR-124a, and the upregulation of BRD4 could potentially reverse the miR-124a effects on AAA cell phenotype. | 1. [49] Liu et al. 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Diktonaite, K.; Qiu, H. Epigenetic Reader Bromodomain-Containing Protein 4 in Aging-Related Vascular Pathologies and Diseases: Molecular Basis, Functional Relevance, and Clinical Potential. Biomolecules 2023, 13, 1135. https://doi.org/10.3390/biom13071135
Zheng X, Diktonaite K, Qiu H. Epigenetic Reader Bromodomain-Containing Protein 4 in Aging-Related Vascular Pathologies and Diseases: Molecular Basis, Functional Relevance, and Clinical Potential. Biomolecules. 2023; 13(7):1135. https://doi.org/10.3390/biom13071135
Chicago/Turabian StyleZheng, Xiaoxu, Kotryna Diktonaite, and Hongyu Qiu. 2023. "Epigenetic Reader Bromodomain-Containing Protein 4 in Aging-Related Vascular Pathologies and Diseases: Molecular Basis, Functional Relevance, and Clinical Potential" Biomolecules 13, no. 7: 1135. https://doi.org/10.3390/biom13071135