



In Silico Analysis of Pyeongwi-San Involved in Inflammatory Bowel Disease Treatment Using Network Pharmacology, Molecular Docking, and Molecular Dynamics



Abstract
:1. Introduction
2. Materials and Methods
2.1. Active Compounds Screening
2.2. Disease-Related Gene Selection
2.3. Target Prediction
2.4. Network Construction
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
2.6. Molecular Docking
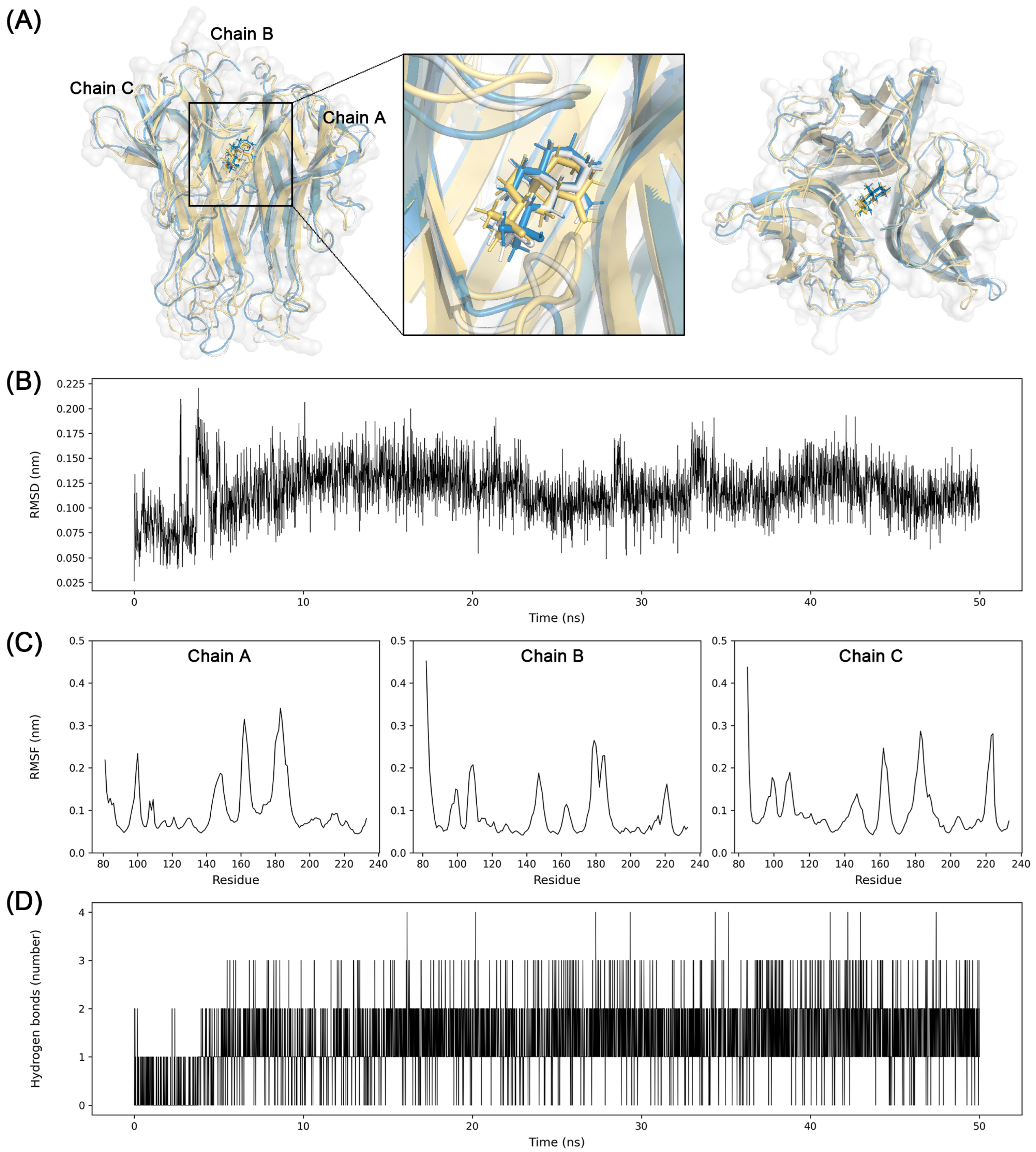
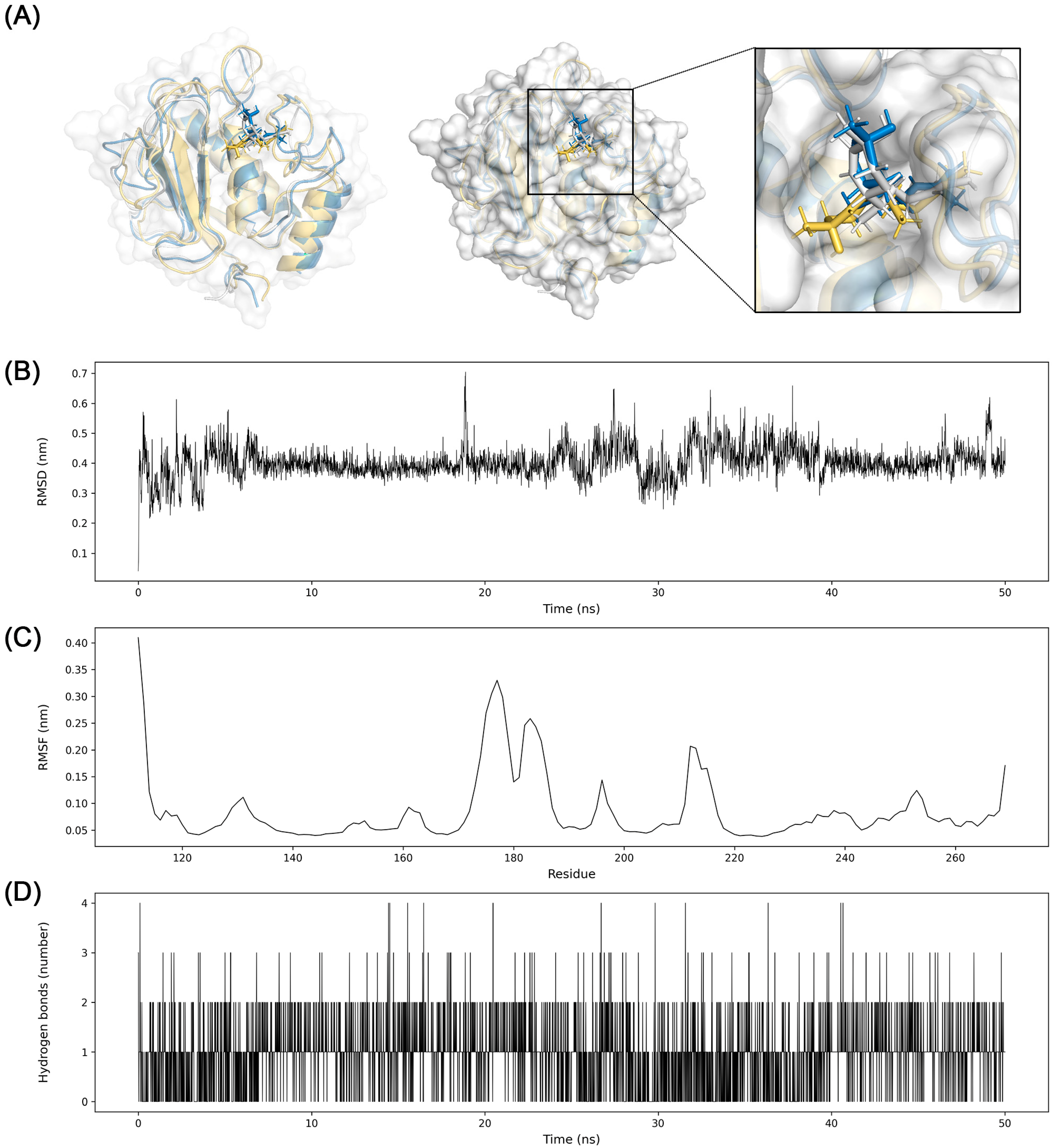
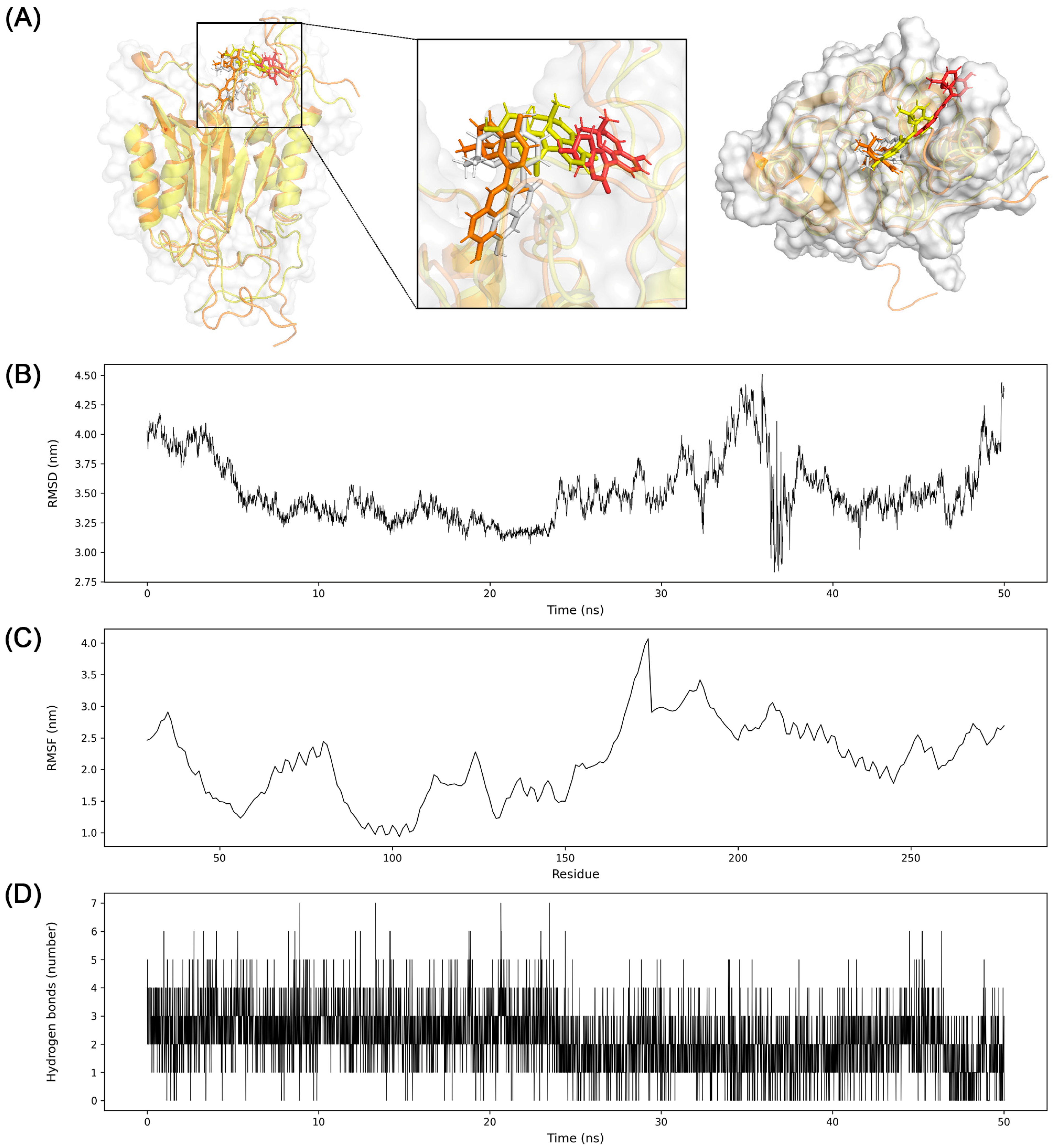
2.7. Molecular Dynamics Simulation
3. Results
3.1. Screening Active Compounds of PWS
3.2. Target Prediction
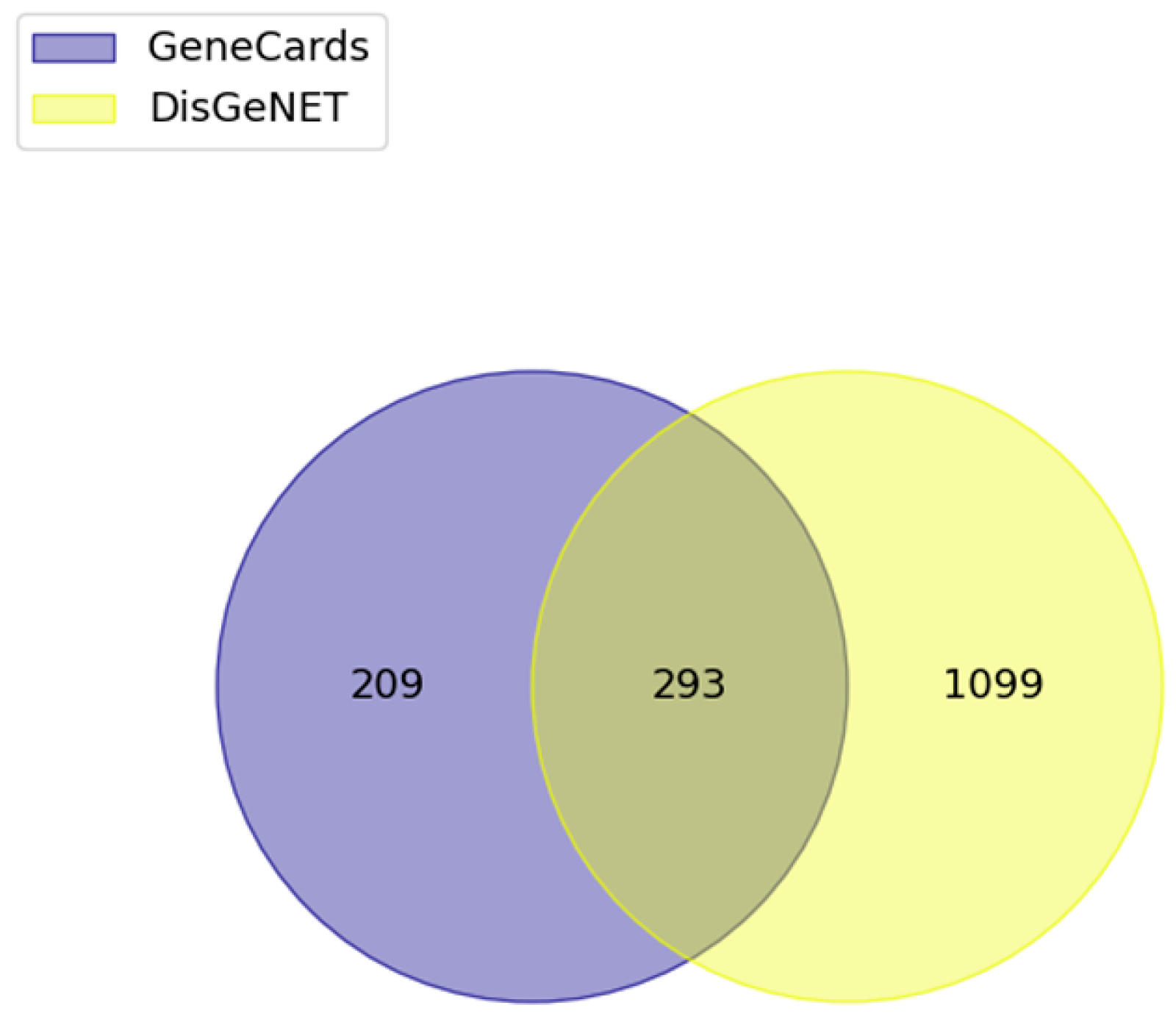
3.3. Disease Gene Association
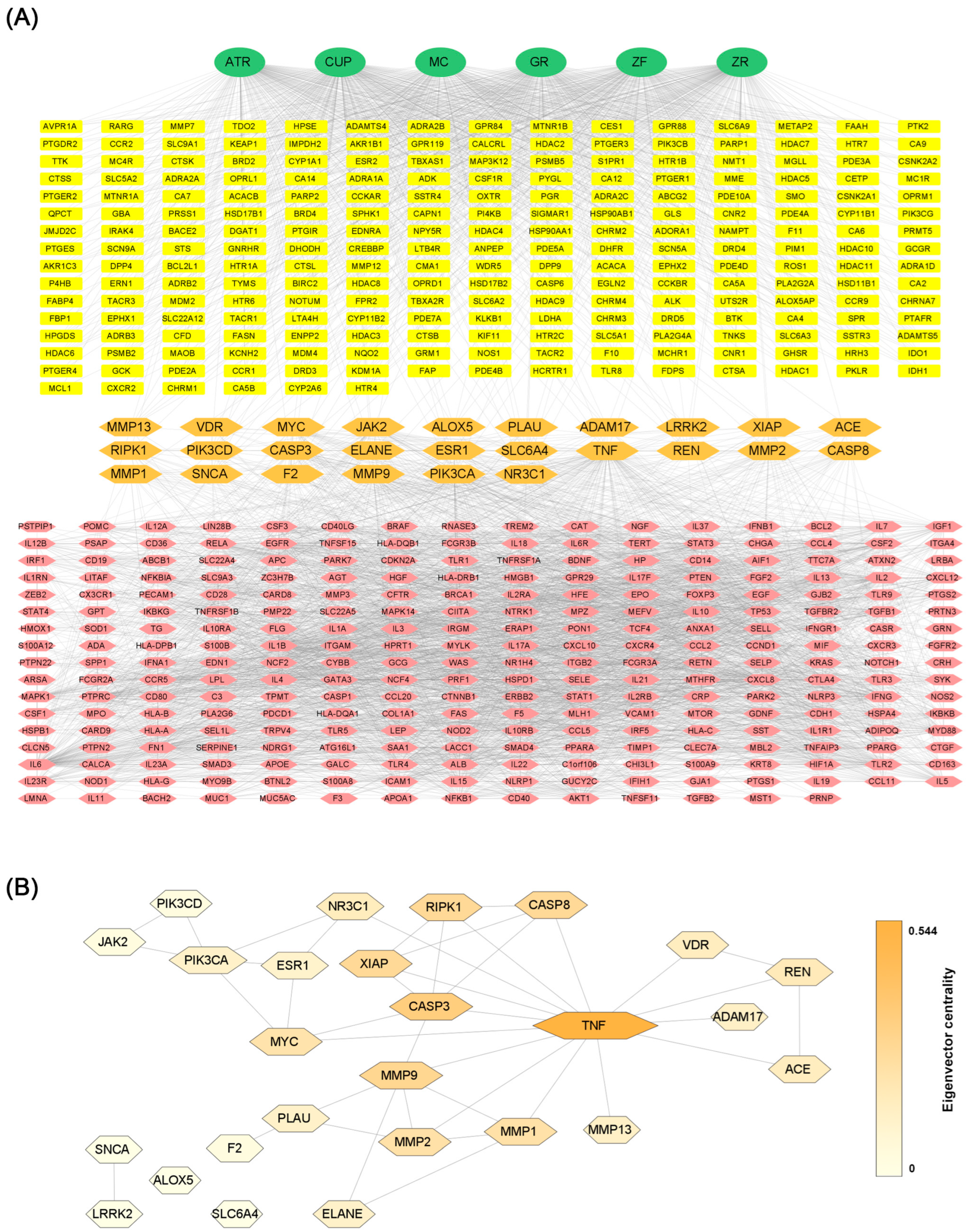
3.4. Network Analysis
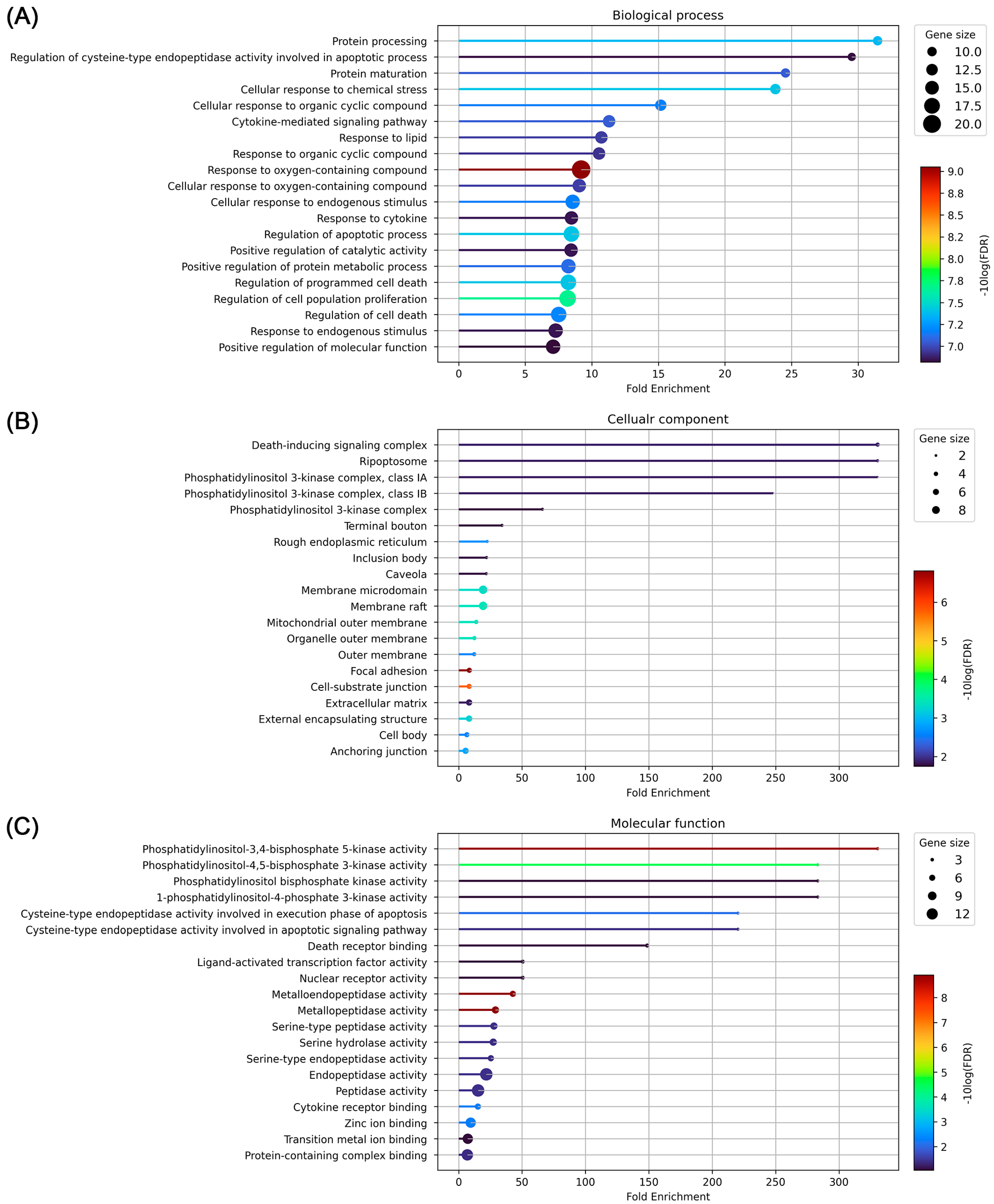
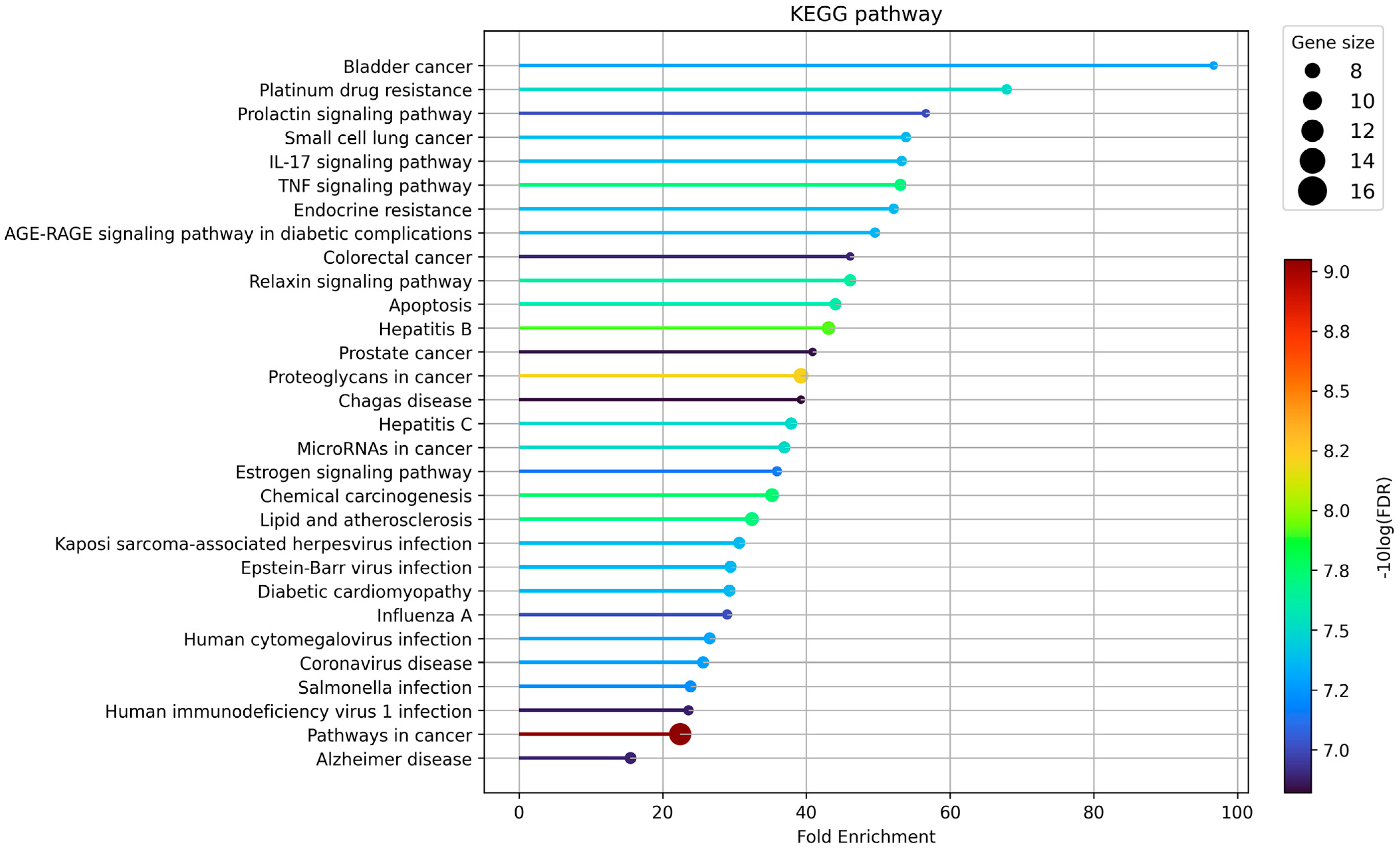
3.5. GO and KEGG Pathway Enrichment Analysis
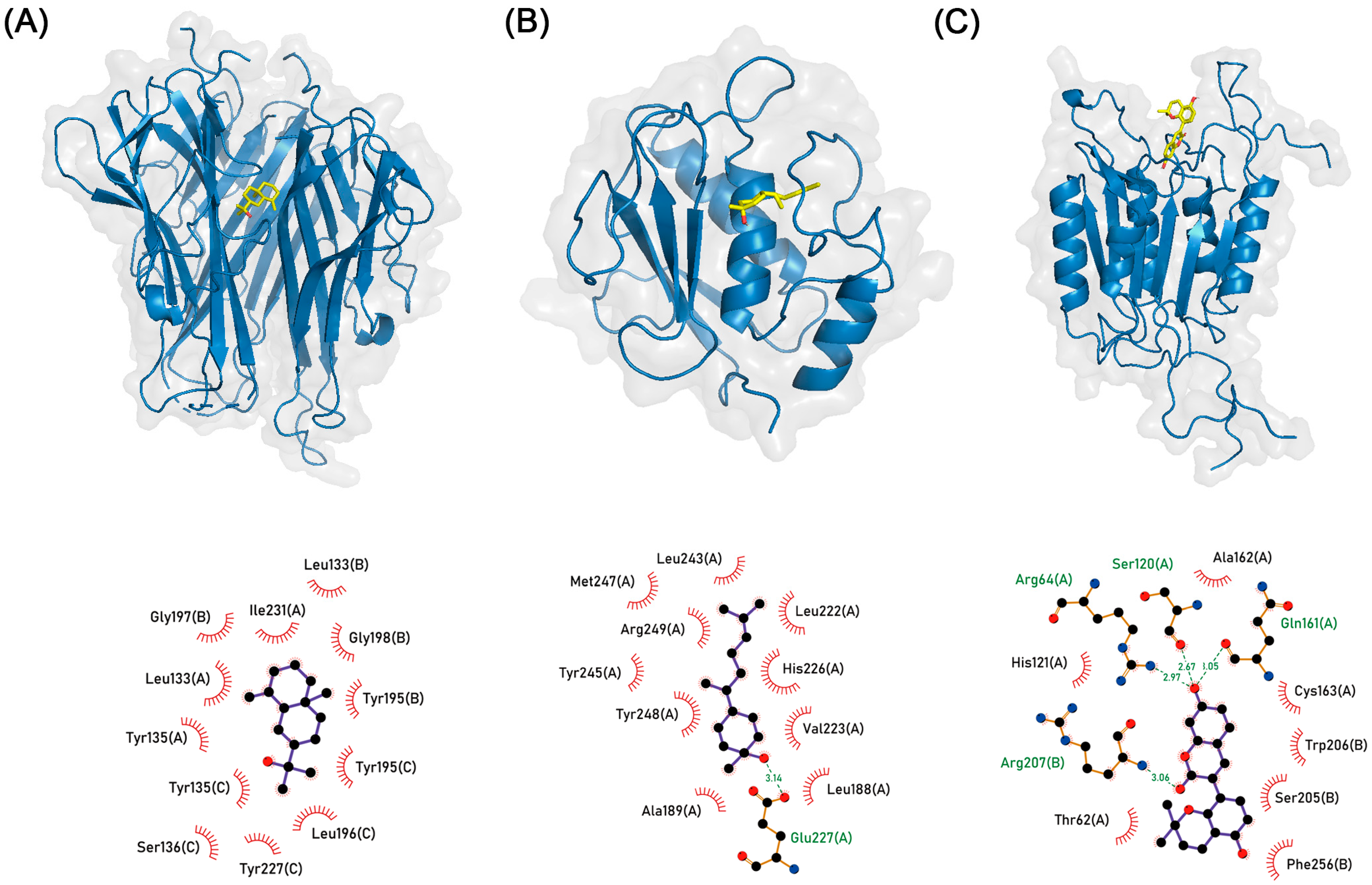
3.6. Molecular Docking Simulation
3.7. Molecular Dynamics Simulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benchimol, E.I.; Fortinsky, K.J.; Gozdyra, P.; Van den Heuvel, M.; Van Limbergen, J.; Griffiths, A.M. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm. Bowel Dis. 2011, 17, 423–439. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology. Nat. Biotechnol. 2007, 25, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wu, T. A Network Pharmacology-Based Study on Vital Pharmacological Pathways and Targets of Eucommiae Cortex Acting on Osteoporosis. Biomed. Res. Int. 2022, 2022, 8510842. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.Y.; Zhang, M.; Cen, H.; Chen, Y.Q.; Wu, Y.F.; Lu, F.; Zhou, J.; Liu, X.S.; Gu, Y.Y. Salvia miltiorrhiza Bunge (Danshen) and Bioactive Compound Tanshinone IIA Alleviates Cisplatin-Induced Acute Kidney Injury Through Regulating PXR/NF-kappaB Signaling. Front. Pharmacol. 2022, 13, 860383. [Google Scholar] [CrossRef]
- Riedlinger, J.E.; Tan, P.W.; Lu, W. Ping wei san, a Chinese medicine for gastrointestinal disorders. Ann. Pharmacother. 2001, 35, 228–235. [Google Scholar] [CrossRef]
- Lee, B.; Ahn, E.K.; Yang, C. Herbal Medicine Prescriptions for Functional Dyspepsia: A Nationwide Population-Based Study in Korea. Evid. Based Complement. Altern. Med. 2022, 2022, 3306420. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Bose, S.; Lim, S.K.; Kim, H. Preventive Effects of Pyungwi-san against Dextran Sulfate Sodium- and Clostridium difficile-Induced Inflammatory Bowel Disease in Mice. Int. J. Mol. Sci. 2019, 20, 6346. [Google Scholar] [CrossRef]
- Cha, J.Y.; Jung, J.Y.; Jung, J.Y.; Lee, J.R.; Cho, I.J.; Ku, S.K.; Byun, S.H.; Ahn, Y.T.; Lee, C.W.; Kim, S.C.; et al. Inhibitory effects of traditional herbal formula pyungwi-san on inflammatory response in vitro and in vivo. Evid. Based Complement. Altern. Med. 2013, 2013, 630198. [Google Scholar] [CrossRef]
- Kim, S.K.; Nam, S.; Jang, H.; Kim, A.; Lee, J.J. TM-MC: A database of medicinal materials and chemical compounds in Northeast Asian traditional medicine. BMC Complement. Altern. Med. 2015, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zhang, X.; Pan, X.; Wang, B.; Ji, C.; Qi, Y.; Zhang, J.Z.H. HobPre: Accurate prediction of human oral bioavailability for small molecules. J. Cheminform. 2022, 14, 1. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Abugessaisa, I., Kasukawa, T., Eds.; Springer: Singapore, 2021; pp. 27–56. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein. Sci. 2016, 86, 2.9.1–2.9.37. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.G. Accuracy and Efficiency of the Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Xue, R.; Fang, Z.; Zhang, M.; Yi, Z.; Wen, C.; Shi, T. TCMID: Traditional Chinese Medicine integrative database for herb molecular mechanism analysis. Nucleic Acids Res. 2013, 41, D1089–D1095. [Google Scholar] [CrossRef]
- Chen, C.Y. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y.; et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminform. 2014, 6, 13. [Google Scholar] [CrossRef]
- Kim, M.T.; Sedykh, A.; Chakravarti, S.K.; Saiakhov, R.D.; Zhu, H. Critical evaluation of human oral bioavailability for pharmaceutical drugs by using various cheminformatics approaches. Pharm. Res. 2014, 31, 1002–1014. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Falcon-Cano, G.; Molina, C.; Cabrera-Perez, M.A. ADME Prediction with KNIME: Development and Validation of a Publicly Available Workflow for the Prediction of Human Oral Bioavailability. J. Chem. Inf. Model. 2020, 60, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef]
- Badkas, A.; De Landtsheer, S.; Sauter, T. Topological network measures for drug repositioning. Brief. Bioinform. 2021, 22, bbaa357. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, G.; Margulies, D.S.; Horstmann, A.; Pleger, B.; Lepsien, J.; Goldhahn, D.; Schloegl, H.; Stumvoll, M.; Villringer, A.; Turner, R. Eigenvector centrality mapping for analyzing connectivity patterns in fMRI data of the human brain. PLoS ONE 2010, 5, e10232. [Google Scholar] [CrossRef] [PubMed]
- Nikkonen, A.; Kolho, K.L. Infliximab and its biosimilar produced similar first-year therapy outcomes in patients with inflammatory bowel disease. Acta Paediatr. 2020, 109, 836–841. [Google Scholar] [CrossRef]
- Busquets, D.; Mas-de-Xaxars, T.; Lopez-Siles, M.; Martinez-Medina, M.; Bahi, A.; Sabat, M.; Louvriex, R.; Miquel-Cusachs, J.O.; Garcia-Gil, J.L.; Aldeguer, X. Anti-tumour Necrosis Factor Treatment with Adalimumab Induces Changes in the Microbiota of Crohn’s Disease. J. Crohn’s Colitis 2015, 9, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Li, X.; Goretsky, T.; Weiss, H.L.; Barrett, T.A.; Gao, T. Loss of PHLPP protects against colitis by inhibiting intestinal epithelial cell apoptosis. Biochim. Biophys. Acta 2015, 1852, 2013–2023. [Google Scholar] [CrossRef]
- Wang, X.; Cui, X.; Zhu, C.; Li, M.; Zhao, J.; Shen, Z.; Shan, X.; Wang, L.; Wu, H.; Shen, Y.; et al. FKBP11 protects intestinal epithelial cells against inflammationinduced apoptosis via the JNKcaspase pathway in Crohn’s disease. Mol. Med. Rep. 2018, 18, 4428–4438. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef]
- Al-Sadi, R.; Youssef, M.; Rawat, M.; Guo, S.; Dokladny, K.; Haque, M.; Watterson, M.D.; Ma, T.Y. MMP-9-induced increase in intestinal epithelial tight permeability is mediated by p38 kinase signaling pathway activation of MLCK gene. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G278–G290. [Google Scholar] [CrossRef]
- Wu, C.; Cao, L.; Liu, M.; Zhang, W.; Chen, H.; Wang, R.; Liu, C.; He, Z. Exploring the mechanisms underlying the therapeutic effect of the drug pair Rhubarb-Coptis in diabetic nephropathy using network pharmacology and molecular docking analysis. Ann. Transl. Med. 2022, 10, 1343. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, F.C.; Li, G. A Network Pharmacology-Based Study of Potential Targets of Angelicae Pubescentis-Herba Taxilli Compound for the Treatment of Osteoarthritis. Comput. Math. Methods Med. 2022, 2022, 4286168. [Google Scholar] [CrossRef]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef]
- Xiao, H.Y.; Li, N.; Duan, J.J.; Jiang, B.; Lu, Z.; Ngu, K.; Tino, J.; Kopcho, L.M.; Lu, H.; Chen, J.; et al. Biologic-like In Vivo Efficacy with Small Molecule Inhibitors of TNFalpha Identified Using Scaffold Hopping and Structure-Based Drug Design Approaches. J. Med. Chem. 2020, 63, 15050–15071. [Google Scholar] [CrossRef]
- O’Connell, J.; Porter, J.; Kroeplien, B.; Norman, T.; Rapecki, S.; Davis, R.; McMillan, D.; Arakaki, T.; Burgin, A.; Fox Iii, D.; et al. Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer. Nat. Commun. 2019, 10, 5795. [Google Scholar] [CrossRef] [PubMed]
- Tandon, A.; Sinha, S. Structural insights into the binding of MMP9 inhibitors. Bioinformation 2011, 5, 310–314. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Kim, S.M.; Yee, S.M.; Kim, E.M.; Lee, E.H.; Choi, H.R.; Lee, Y.S.; Yang, W.K.; Kim, H.Y.; Kim, K.H.; et al. Daucosterol suppresses dextran sulfate sodium (DSS)-induced colitis in mice. Int. Immunopharmacol. 2019, 72, 124–130. [Google Scholar] [CrossRef]
- Tang, X.; Huang, G.; Zhang, T.; Li, S. Elucidation of colon-protective efficacy of diosgenin in experimental TNBS-induced colitis: Inhibition of NF-kappaB/IkB-alpha and Bax/Caspase-1 signaling pathways. Biosci. Biotechnol. Biochem. 2020, 84, 1903–1912. [Google Scholar] [CrossRef]
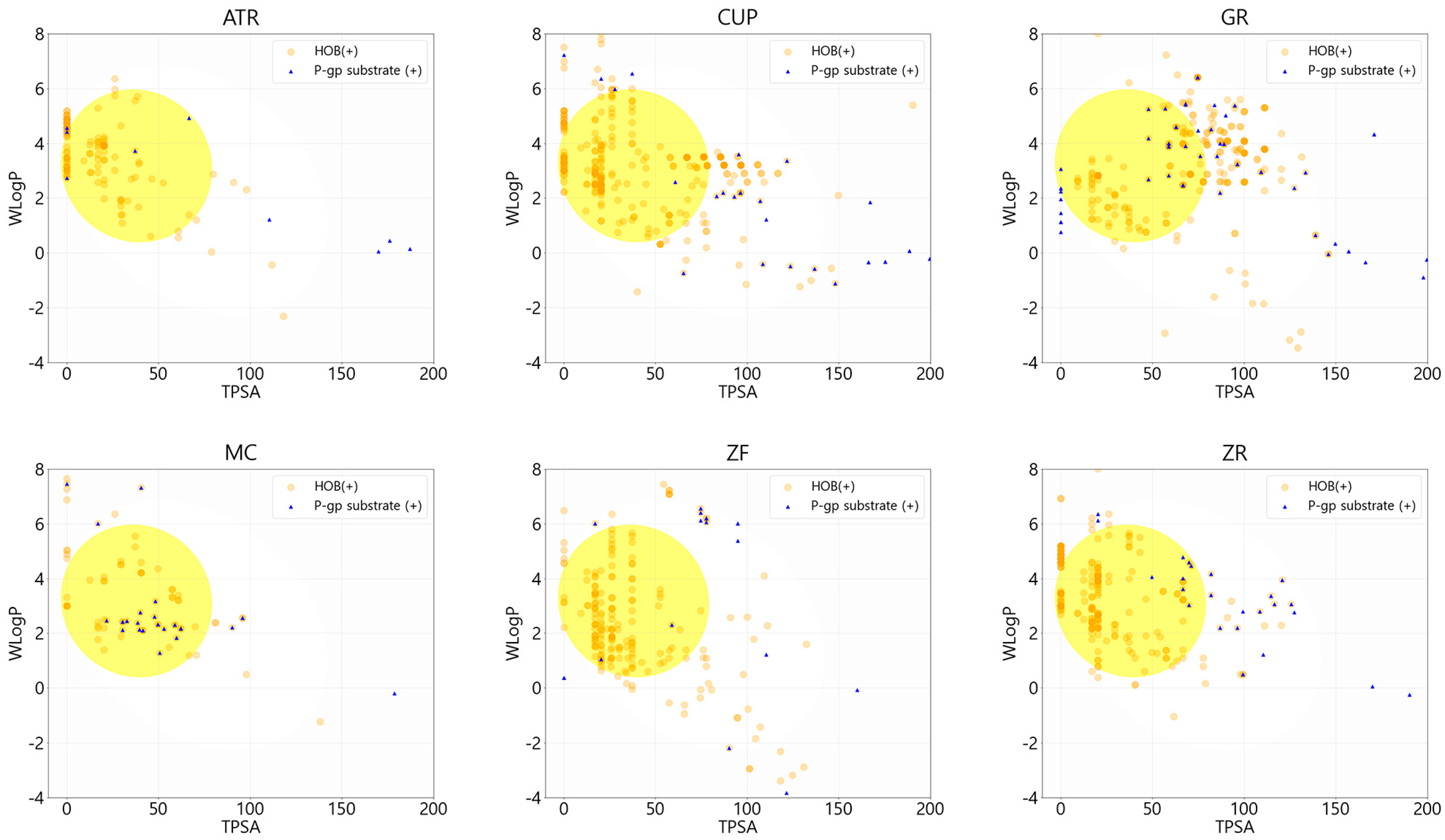
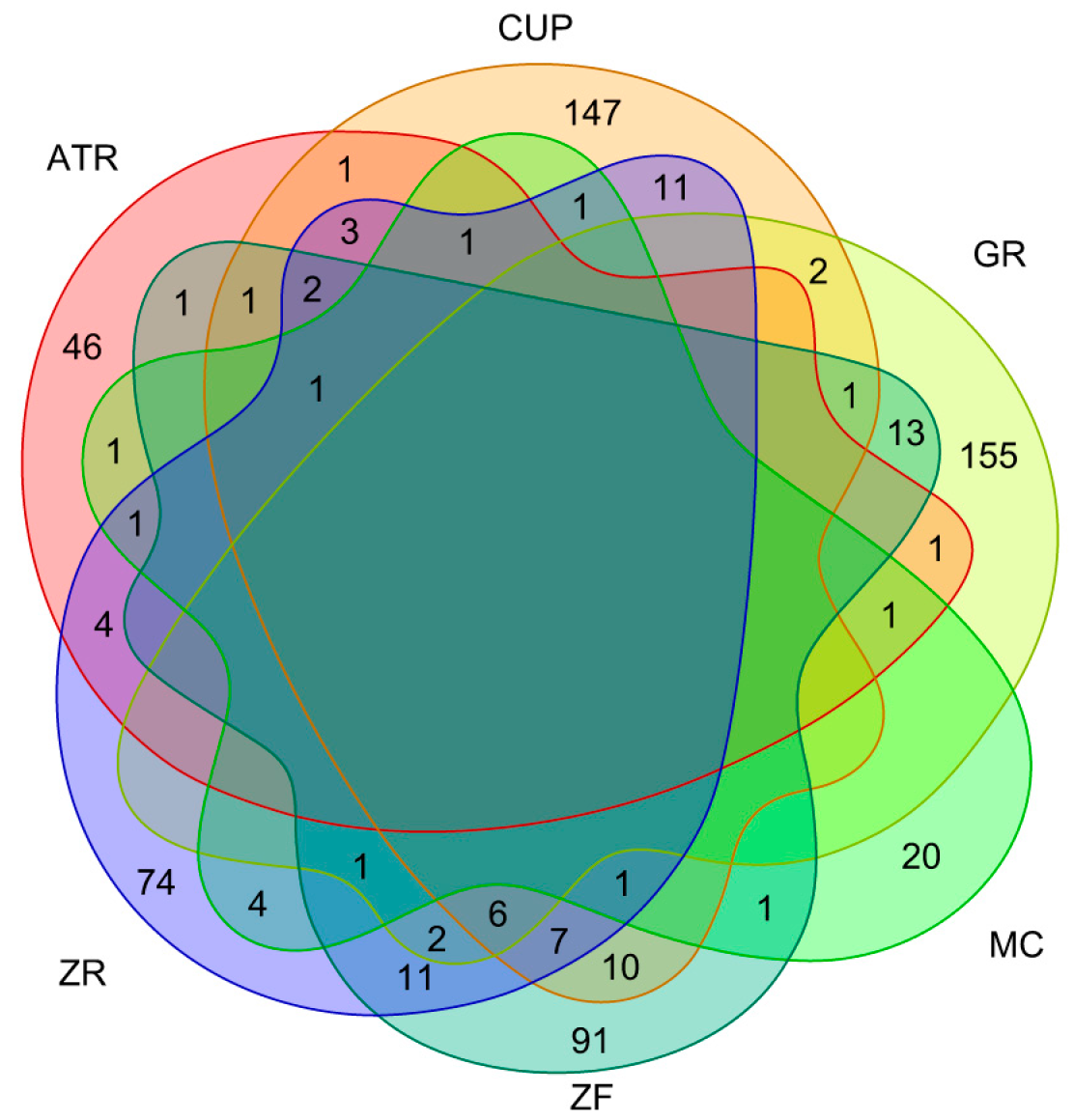
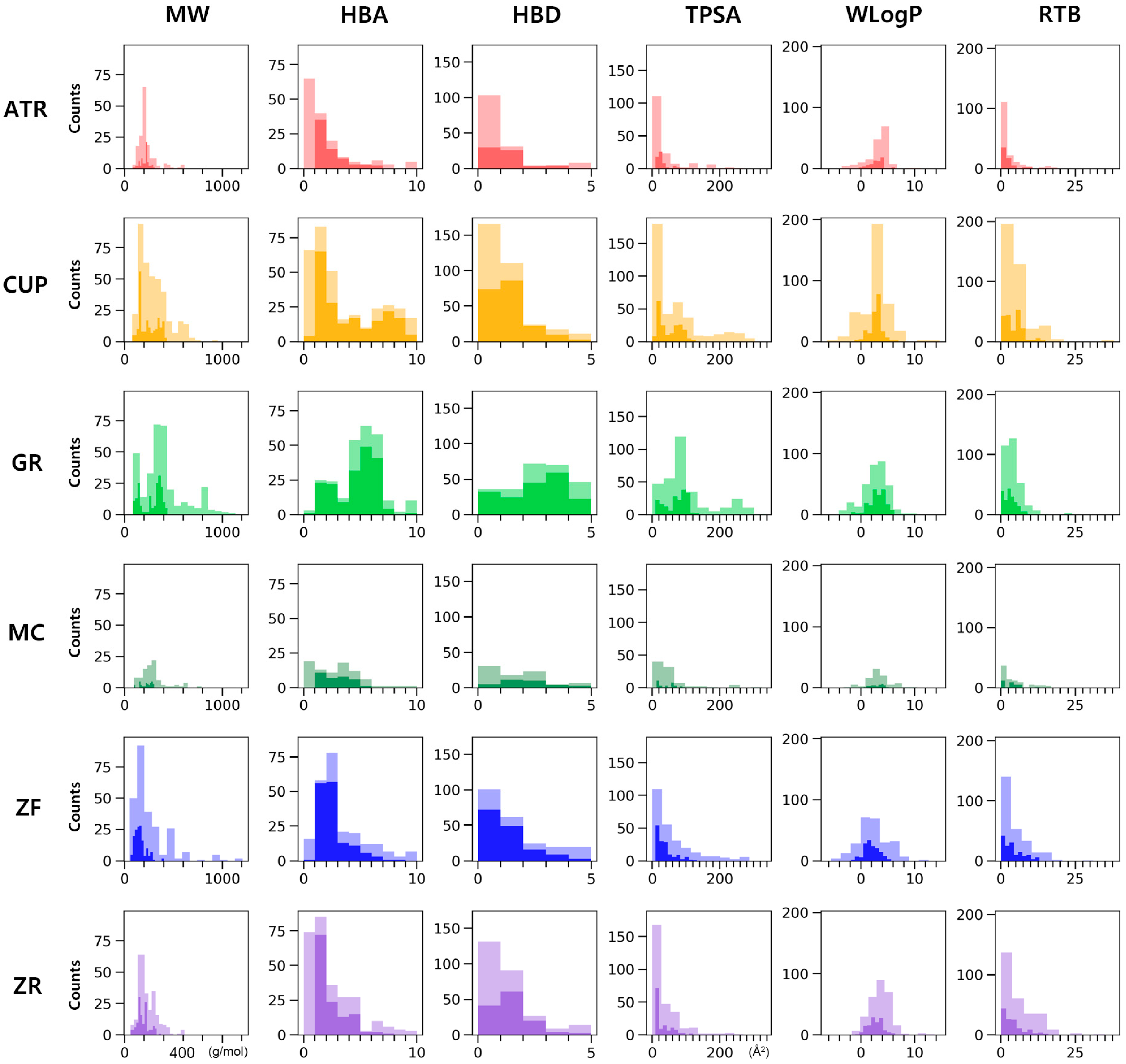
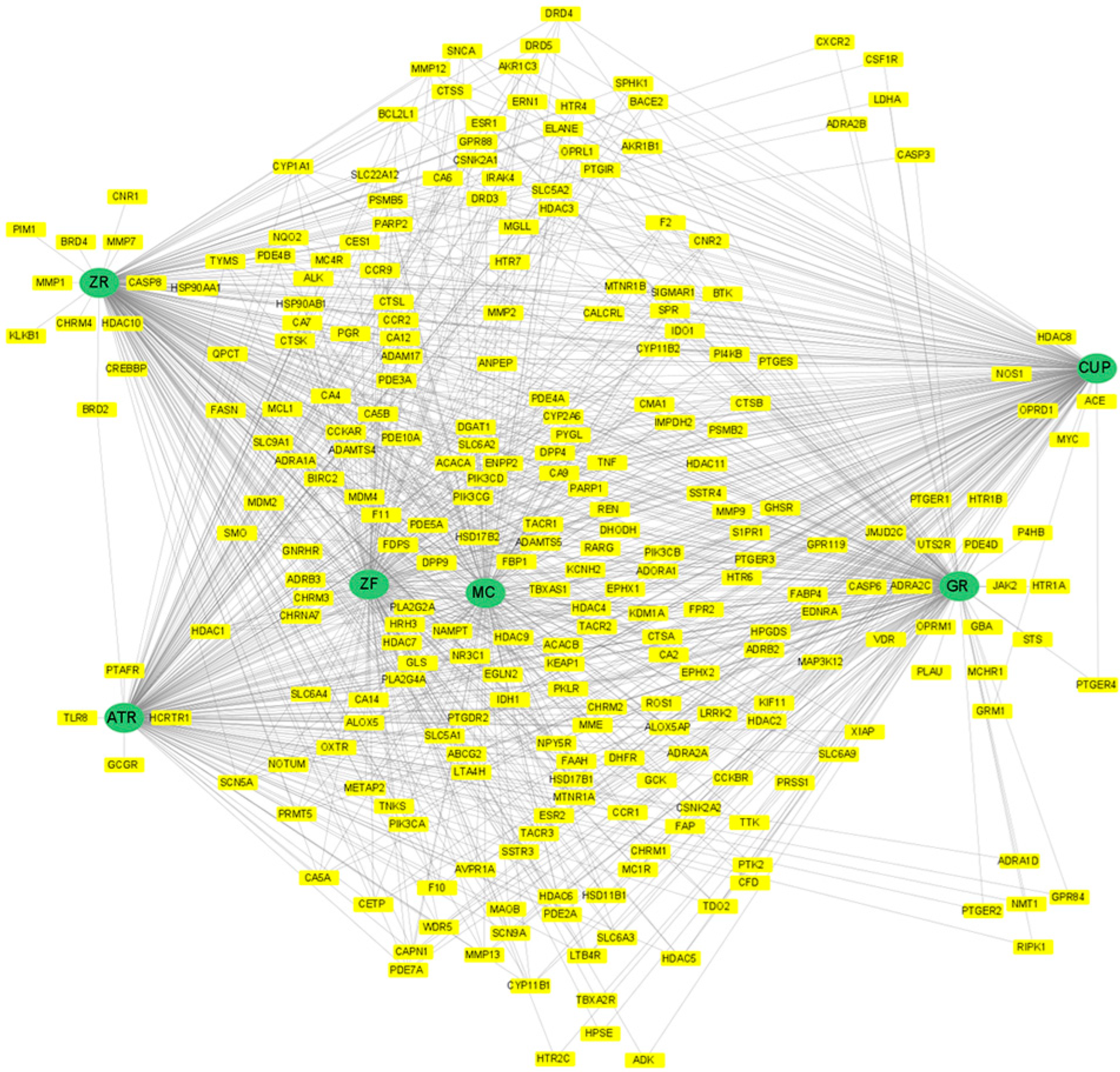

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Degree Centrality | Eigenvector Centrality | Betweenness Centrality | Closeness Centrality |
|---|---|---|---|---|
| TNF (Tumor Necrosis Factor) | 14 | 0.544 | 293.2 | 0.185 |
| CASP3 (Caspase 3) | 6 | 0.353 | 30.5 | 0.175 |
| MMP9 (Matrix Metallopeptidase 9) | 6 | 0.289 | 66.4 | 0.171 |
| PIK3CA (Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha) | 5 | 0.091 | 76.7 | 0.157 |
| MMP2 (Matrix Metallopeptidase 2) | 4 | 0.217 | 27.7 | 0.169 |
| XIAP (X-Linked Inhibitor Of Apoptosis) | 4 | 0.274 | 0.0 | 0.164 |
| CASP8 (Caspase 8) | 4 | 0.274 | 0.0 | 0.164 |
| RIPK1 (Receptor Interacting Serine/Threonine Kinase 1) | 4 | 0.274 | 0.0 | 0.164 |
| MYC (MYC Proto-Oncogene, BHLH Transcription Factor) | 4 | 0.204 | 75.7 | 0.171 |
| MMP1 (Matrix Metallopeptidase 1) | 4 | 0.217 | 13.9 | 0.168 |
| ESR1 (Estrogen Receptor 1) | 3 | 0.084 | 0.7 | 0.155 |
| NR3C1 (Nuclear Receptor Subfamily 3 Group C Member 1) | 3 | 0.137 | 52.3 | 0.170 |
| REN (Renin) | 3 | 0.153 | 1.0 | 0.163 |
| PLAU (Plasminogen Activator, Urokinase) | 3 | 0.099 | 40.0 | 0.153 |
| VDR (Vitamin D Receptor) | 2 | 0.132 | 0.0 | 0.162 |
| PIK3CD (Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Delta) | 2 | 0.023 | 0.0 | 0.140 |
| ELANE (Elastase, Neutrophil Expressed) | 2 | 0.096 | 0.0 | 0.152 |
| ACE (Angiotensin I Converting Enzyme) | 2 | 0.132 | 0.0 | 0.162 |
| JAK2 (Janus Kinase 2) | 2 | 0.023 | 0.0 | 0.140 |
| ADAM17 (ADAM Metallopeptidase Domain 17) | 1 | 0.103 | 0.0 | 0.161 |
| F2 (Coagulation Factor II, Thrombin) | 1 | 0.019 | 0.0 | 0.137 |
| LRRK2 (Leucine Rich Repeat Kinase 2) | 1 | 0.000 | 0.0 | 0.040 |
| MMP13 (Matrix Metallopeptidase 13) | 1 | 0.103 | 0.0 | 0.161 |
| SNCA (Synuclein Alpha) | 1 | 0.000 | 0.0 | 0.040 |
| SLC6A4 (Solute Carrier Family 6 Member 4) | 0 | 0.000 | 0.0 | 0.038 |
| ALOX5 (Arachidonate 5-Lipoxygenase) | 0 | 0.000 | 0.0 | 0.038 |
| Medicine | Compound | PubChem ID | Target Protein (PDB ID) | Binding Affinity (kcal/mol) |
|---|---|---|---|---|
| ZR | β-eudesmol | 91457 | TNF (7JRA) | −8.259 |
| ZR | (3R,6R,7S)-1,10-bisaboladien-3-ol | 71813358 | MMP9 (4WZV) | −8.131 |
| GR | Glabrocoumarin | 11427657 | CASP3 (2C2M) | −8.238 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, C.-H.; Kim, H.-Y.; Seo, J.E.; Lee, H.; Kim, S. In Silico Analysis of Pyeongwi-San Involved in Inflammatory Bowel Disease Treatment Using Network Pharmacology, Molecular Docking, and Molecular Dynamics. Biomolecules 2023, 13, 1322. https://doi.org/10.3390/biom13091322
Bae C-H, Kim H-Y, Seo JE, Lee H, Kim S. In Silico Analysis of Pyeongwi-San Involved in Inflammatory Bowel Disease Treatment Using Network Pharmacology, Molecular Docking, and Molecular Dynamics. Biomolecules. 2023; 13(9):1322. https://doi.org/10.3390/biom13091322
Chicago/Turabian StyleBae, Chang-Hwan, Hee-Young Kim, Ji Eun Seo, Hanul Lee, and Seungtae Kim. 2023. "In Silico Analysis of Pyeongwi-San Involved in Inflammatory Bowel Disease Treatment Using Network Pharmacology, Molecular Docking, and Molecular Dynamics" Biomolecules 13, no. 9: 1322. https://doi.org/10.3390/biom13091322
APA StyleBae, C.-H., Kim, H.-Y., Seo, J. E., Lee, H., & Kim, S. (2023). In Silico Analysis of Pyeongwi-San Involved in Inflammatory Bowel Disease Treatment Using Network Pharmacology, Molecular Docking, and Molecular Dynamics. Biomolecules, 13(9), 1322. https://doi.org/10.3390/biom13091322