
Comprehensive Organ-Specific Profiling of Douglas Fir (Pseudotsuga menziesii) Proteome

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Sampling
2.2. Sample Preparation and Protein Digestion
2.3. nLC-MS/MS Analysis and Label-Free Quantitative Data Analysis
2.4. Database Search and Results Processing
2.5. Gene Ontology (GO) Analysis
2.6. Statistical Analysis
3. Results
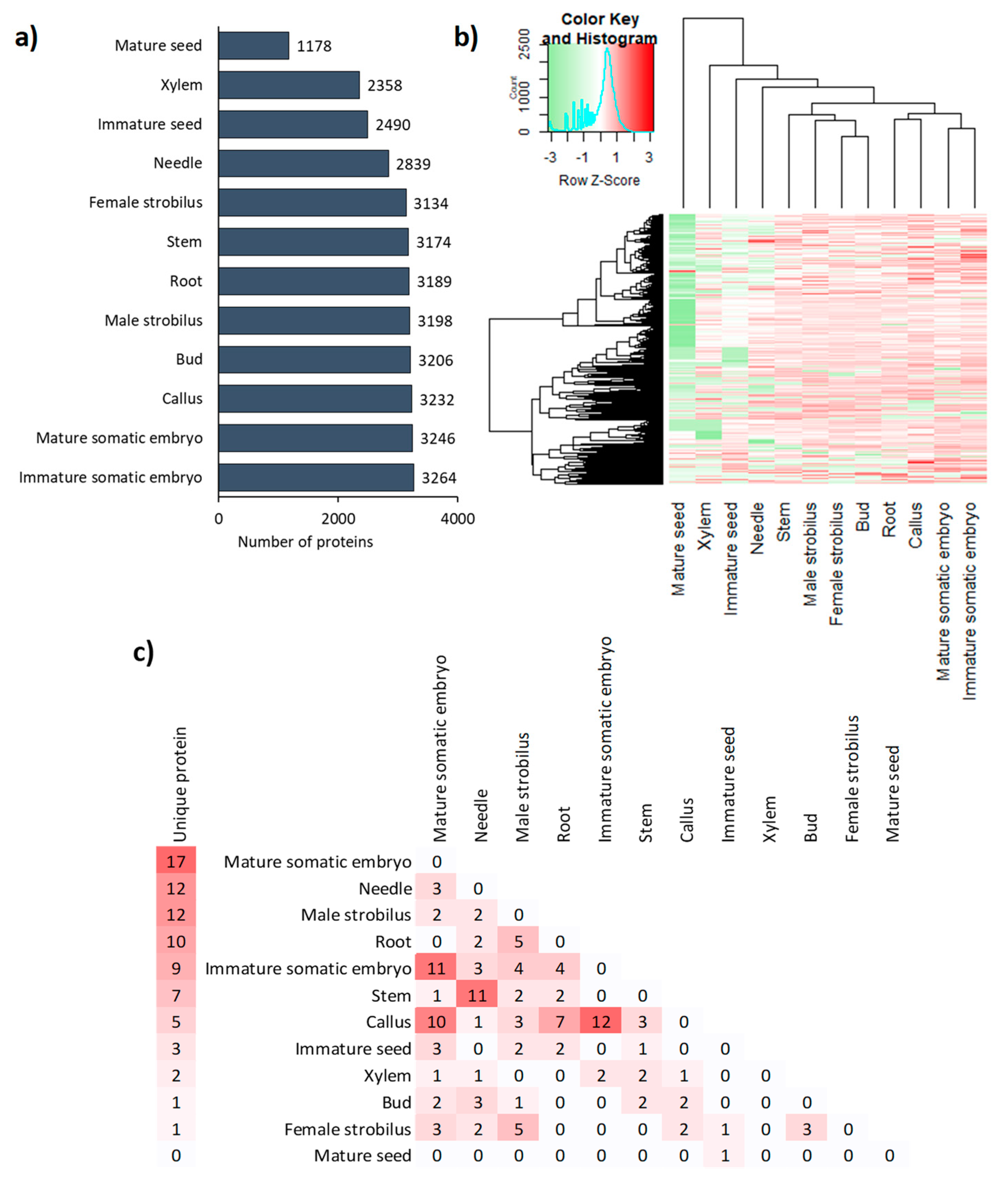
3.1. A wide Diversity and Abundance of Proteins Observed in All Organs
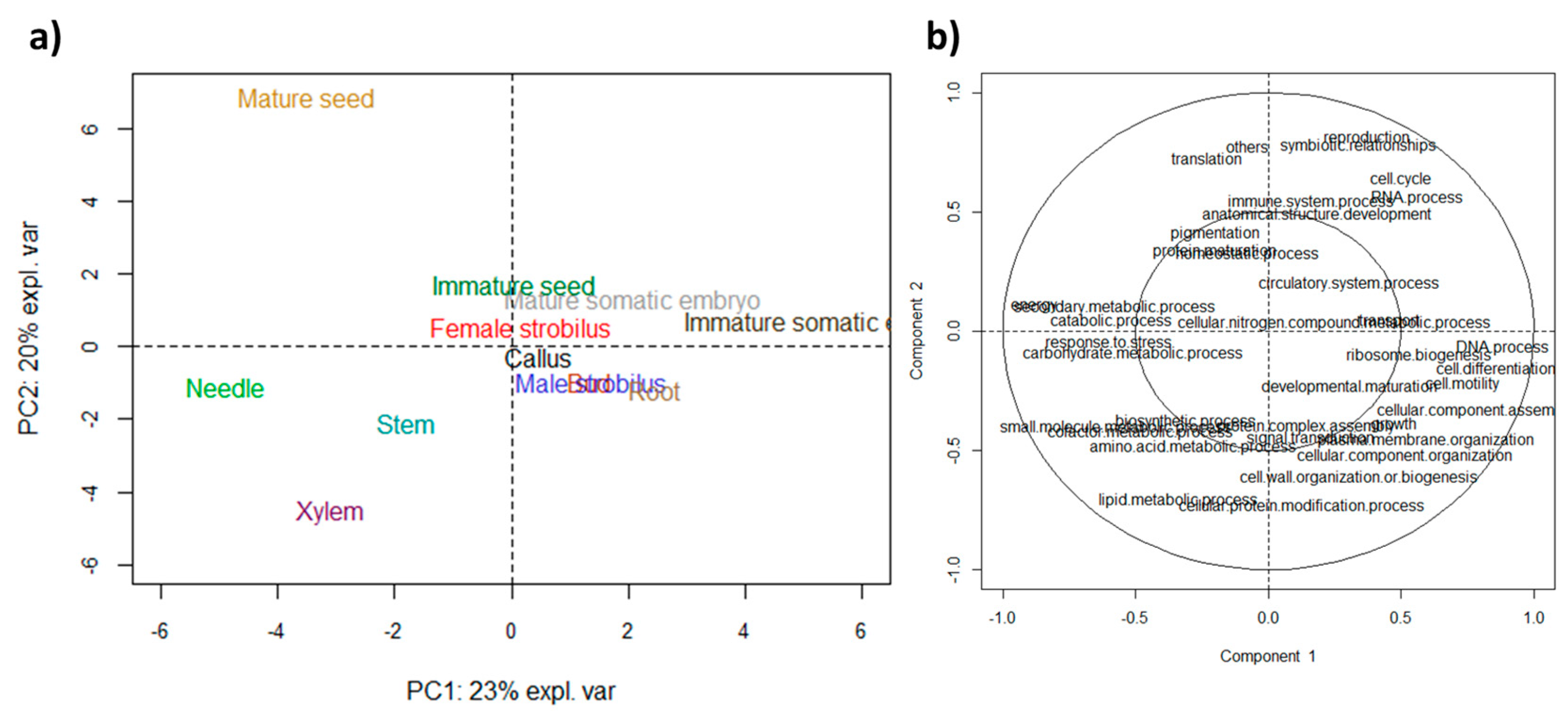
3.2. Functional Specificities were Highlighted by GO Analyses
3.3. First Analysis of Active Biological Processes in Douglas Fir Organs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bastien, J.-C.; Sanchez, L.; Michaud, D. Douglas-Fir (Pseudotsuga menziesii (Mirb.) Franco). In Forest Tree Breeding in Europe. Current State-of-the-Art and Perspectives; Pâques, L.E., Ed.; Springer: Dordrecht, The Netherlands; Berlin/Heidelberg, Germany; New York, NY, USA; London, UK, 2013; pp. 325–369. [Google Scholar]
- Hagerman, S.M.; Pelai, R. Responding to climate change in forest management: Two decades of recommendations. Front. Ecol. Environ. 2018, 16, 579–587. [Google Scholar] [CrossRef]
- Pyhäjärvi, T.; Kujala, S.T.; Savolainen, O. 275 years of forestry meets genomics in Pinus sylvestris. Evol. Appl. 2020, 13, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Wakamiya, I.; Newton, R.J.; Johnston, J.S.; Price, H.J. Genome size and environmental factors in the genus Pinus. Am. J. Bot. 1993, 80, 1235–1241. [Google Scholar] [CrossRef]
- Stival Sena, J.; Giguère, I.; Boyle, B.; Rigault, P.; Birol, I.; Zuccolo, A.; Ritland, K.; Ritland, C.; Bohlmann, J.; Jones, S.; et al. Evolution of gene structure in the conifer Picea glauca: A comparative analysis of the impact of intron size. BMC Plant Biol. 2014, 14, 95. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Leitch, I.J. The plant DNA C-values database (release 7.1): An updated online repository of plant genome size data for comparative studies. New Phytol. 2020, 226, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Kovach, A.; Wegrzyn, J.L.; Parra, G.; Holt, C.; Bruening, G.E.; Loopstra, C.A.; Hartigan, J.; Yandell, M.; Langley, C.H.; Korf, I.; et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genom. 2010, 11, 420. [Google Scholar] [CrossRef] [PubMed]
- Morse, A.M.; Peterson, D.G.; Islam-Faridi, M.N.; Smith, K.E.; Magbanua, Z.; Garcia, S.A.; Kubisiak, T.L.; Amerson, H.V.; Carlson, J.E.; Nelson, C.D.; et al. Evolution of genome size and complexity in Pinus. PLoS ONE 2009, 4, e4332. [Google Scholar] [CrossRef]
- Wan, T.; Gong, Y.; Liu, Z.; Zhou, Y.; Dai, C.; Wang, Q. Evolution of complex genome architecture in gymnosperms. GigaScience 2022, 11, giac078. [Google Scholar] [CrossRef]
- Mackay, J.; Dean, J.F.D.; Plomion, C.; Peterson, D.G.; Cánovas, F.M.; Pavy, N.; Ingvarsson, P.K.; Savolainen, O.; Guevara, M.Á.; Fluch, S.; et al. Towards decoding the conifer giga-genome. Plant Mol. Biol. 2012, 80, 555–569. [Google Scholar] [CrossRef]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef]
- Rellstab, C.; Dauphin, B.; Zoller, S.; Brodbeck, S.; Gugerli, F. Using transcriptome sequencing and pooled exome capture to study local adaptation in the giga-genome of Pinus cembra. Mol. Ecol. Resour. 2019, 19, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.-H.; Li, Z.-X.; Yuan, H.-W.; Chen, X.-Y.; Li, Y.; Li, W. Transcriptome characterisation of Pinus tabuliformis and evolution of genes in the Pinus phylogeny. BMC Genom. 2013, 14, 263. [Google Scholar] [CrossRef] [PubMed]
- Pinosio, S.; González-Martínez, S.C.; Bagnoli, F.; Cattonaro, F.; Grivet, D.; Marroni, F.; Lorenzo, Z.; Pausas, J.G.; Verdú, M.; Vendramin, G.G. First insights into the transcriptome and development of new genomic tools of a widespread circum-Mediterranean tree species, Pinus halepensis Mill. Mol. Ecol. Resour. 2014, 14, 846–856. [Google Scholar] [CrossRef]
- Hall, D.E.; Yuen, M.M.S.; Jancsik, S.; Quesada, A.L.; Dullat, H.K.; Li, M.; Henderson, H.; Arango-Velez, A.; Liao, N.Y.; Docking, R.T.; et al. Transcriptome resources and functional characterization of monoterpene synthases for two host species of the mountain pine beetle, lodgepole pine (Pinus contorta) and jack pine (Pinus banksiana). BMC Plant Biol. 2013, 13, 80. [Google Scholar] [CrossRef]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-fir genome sequence reveals specialization of the photosynthetic apparatus in Pinaceae. G3 Genes Genomes Genet. 2017, 7, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.J.; Fahrenkrog, A.M.; Neves, L.G.; Resende, M.F.R.; Dervinis, C.; Davis, J.M.; Holliday, J.A.; Kirst, M. Exome resequencing reveals evolutionary history, genomic diversity, and targets of selection in the conifers Pinus taeda and Pinus elliottii. Genome Biol. Evol. 2019, 11, 508–520. [Google Scholar] [CrossRef]
- Lu, M.; Krutovsky, K.V.; Nelson, C.D.; Koralewski, T.E.; Byram, T.D.; Loopstra, C.A. Exome genotyping, linkage disequilibrium and population structure in loblolly pine (Pinus taeda L.). BMC Genom. 2016, 17, 730. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef]
- Castillejo, M.A.; Pascual, J.; Jorrin-Novo, J.V.; Balbuena, T.S. Proteomics research in forest trees: A 2012–2022 update. Front. Plant Sci. 2023, 14, 1130665. [Google Scholar] [CrossRef] [PubMed]
- Gion, J.-M.; Lalanne, C.; Le Provost, G.; Ferry-Dumazet, H.; Paiva, J.; Chaumeil, P.; Frigerio, J.-M.; Brach, J.; Barré, A.; de Daruvar, A.; et al. The proteome of maritime pine wood forming tissue. Proteomics 2005, 5, 3731–3751. [Google Scholar] [CrossRef]
- Abril, N.; Gion, J.-M.; Kerner, R.; Müller-Starck, G.; Cerrillo, R.M.N.; Plomion, C.; Renaut, J.; Valledor, L.; Jorrin-Novo, J.V. Proteomics research on forest trees, the most recalcitrant and orphan plant species. Phytochemistry 2011, 72, 1219–1242. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.-D.; Castillejo, M.Á.; Sánchez-Lucas, R.; Guerrero-Sanchez, V.M.; López-Hidalgo, C.; Romero-Rodríguez, C.; Valero-Galván, J.; Sghaier-Hammami, B.; Simova-Stoilova, L.; Echevarría-Zomeño, S.; et al. Proteomics, holm oak (Quercus ilex L.) and other recalcitrant and orphan forest tree species: How do they see each other? Int. J. Mol. Sci. 2019, 20, 692. [Google Scholar] [CrossRef] [PubMed]
- Min, C.W.; Gupta, R.; Agrawal, G.K.; Rakwal, R.; Kim, S.T. Concepts and strategies of soybean seed proteomics using the shotgun proteomics approach. Expert Rev. Proteom. 2019, 16, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Shotgun proteomics as a powerful tool for the study of the proteomes of plants, their pathogens, and plant-pathogen interactions. Proteomes 2022, 10, 5. [Google Scholar] [CrossRef]
- de Francisco, L.R.; Romero-Rodríguez, M.C.; Navarro-Cerrillo, R.M.; Miniño, V.; Perdomo, O.; Jorrín-Novo, J.V. Characterization of the orthodox Pinus occidentalis seed and pollen proteomes by using complementary gel-based and gel-free approaches. J. Proteom. 2016, 143, 382–389. [Google Scholar] [CrossRef]
- Aguilar-Hernández, V.; Loyola-Vargas, V.M. Advanced proteomic approaches to elucidate somatic embryogenesis. Front. Plant Sci. 2018, 9, 1658. [Google Scholar] [CrossRef]
- Prior, N.; Little, S.A.; Boyes, I.; Griffith, P.; Husby, C.; Pirone-Davies, C.; Stevenson, D.W.; Tomlinson, P.B.; von Aderkas, P. Complex reproductive secretions occur in all extant gymnosperm lineages: A proteomic survey of gymnosperm pollination drops. Plant Reprod. 2019, 32, 153–166. [Google Scholar] [CrossRef]
- Guo, Y.T.; Niu, S.H.; El-Kassaby, Y.A.; Li, W. Transcriptomic and proteomic analyses of far-red light effects in inducing shoot elongation in the presence or absence of paclobutrazol in Chinese pine. J. For. Res. 2022, 33, 1033–1043. [Google Scholar] [CrossRef]
- Alfonso, O.B.; Ariza-Mateos, D.; Palacios-Rodríguez, G.; Ginhas Manuel, A.; Ruiz-Gómez, F.J. Seed protein profile of Pinus greggii and Pinus patula through functional genomics analysis. Bosque 2020, 41, 333–344. [Google Scholar] [CrossRef]
- Romero-Rodríguez, M.C.; Pascual, J.; Valledor, L.; Jorrín-Novo, J. Improving the quality of protein identification in non-model species. Characterization of Quercus ilex seed and Pinus radiata needle proteomes by using SEQUEST and custom databases. J. Proteom. 2014, 105, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cao, D.; Chen, Y.; Wei, D.; Wang, Y.; Stevenson, R.A.; Zhu, Y.; Lin, J. Gene expression and proteomic analysis of shoot apical meristem transition from dormancy to activation in Cunninghamia lanceolata (Lamb.) Hook. Sci. Rep. 2016, 6, 19938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, H.; Fu, S.; Chen, B.; Sun, W.; Zhang, J.; Zhang, J. An iTRAQ-based proteomics approach to clarify the molecular physiology of somatic embryo development in Prince Rupprecht’s larch (Larix principis-rupprechtii Mayr). PLoS ONE 2015, 10, e0119987. [Google Scholar] [CrossRef]
- Lamelas, L.; Valledor, L.; Escandón, M.; Pinto, G.; Cañal, M.J.; Meijón, M. Integrative analysis of the nuclear proteome in Pinus radiata reveals thermopriming coupled to epigenetic regulation. J. Exp. Bot. 2020, 71, 2040–2057. [Google Scholar] [CrossRef] [PubMed]
- Duruflé, H.; Clemente, H.S.; Balliau, T.; Zivy, M.; Dunand, C.; Jamet, E. Cell wall proteome analysis of Arabidopsis thaliana mature stems. Proteomics 2017, 17, 1600449. [Google Scholar] [CrossRef] [PubMed]
- López-Hidalgo, C.; Guerrero-Sánchez, V.M.; Gómez-Gálvez, I.; Sánchez-Lucas, R.; Castillejo-Sánchez, M.A.; Maldonado-Alconada, A.M.; Valledor, L.; Jorrín-Novo, J.V. A multi-omics analysis pipeline for the metabolic pathway reconstruction in the orphan species Quercus ilex. Front. Plant Sci. 2018, 9, 935. [Google Scholar] [CrossRef]
- Lelu-Walter, M.-A.; Gautier, F.; Eliášová, K.; Sanchez, L.; Teyssier, C.; Lomenech, A.-M.; Le Metté, C.; Hargreaves, C.; Trontin, J.-F.; Reeves, C. High gellan gum concentration and secondary somatic embryogenesis: Two key factors to improve somatic embryo development in Pseudotsuga menziesii [Mirb.]. Plant Cell Tiss. Organ. Cult. 2018, 132, 137–155. [Google Scholar] [CrossRef]
- Teyssier, C.; Grondin, C.; Bonhomme, L.; Lomenech, A.-M.; Vallance, M.; Morabito, D.; Label, P.; Lelu-Walter, M.-A. Increased gelling agent concentration promotes somatic embryo maturation in hybrid larch (Larix × eurolepsis): A 2-DE proteomic analysis. Physiol. Plant. 2011, 141, 152–165. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Meth. 2007, 4, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Casimiro-Soriguer, C.S.; Muñoz-Mérida, A.; Pérez-Pulido, A.J. Sma3s: A universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics 2017, 17, 1700071. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, F.; Xiao, S.; Jiang, C.; Yu, T.; Chen, L.; Ke, X.; Zhong, Q.; Cheng, Z.; Li, W. Proteomic analysis of the rice (Oryza officinalis) provides clues on molecular tagging of proteins for brown planthopper resistance. BMC Plant Biol. 2019, 19, 30. [Google Scholar] [CrossRef]
- Ramírez-Sánchez, O.; Pérez-Rodríguez, P.; Delaye, L.; Tiessen, A. Plant proteins are smaller because they are encoded by fewer Exons than animal proteins. Genom. Proteom. Bioinform. 2016, 14, 357–370. [Google Scholar] [CrossRef]
- Patole, C.; Bindschedler, L.V. Chapter 4—Plant proteomics: A guide to improve the proteome coverage. In Advances in Biological Science Research; Meena, S.N., Naik, M.M., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 45–67. [Google Scholar]
- Obudulu, O.; Bygdell, J.; Sundberg, B.; Moritz, T.; Hvidsten, T.R.; Trygg, J.; Wingsle, G. Quantitative proteomics reveals protein profiles underlying major transitions in aspen wood development. BMC Genom. 2016, 17, 119. [Google Scholar] [CrossRef]
- Lippert, D.; Jun, Z.; Ralph, S.; Ellis, D.E.; Gilbert, M.; Olafson, R.; Ritland, K.; Ellis, B.; Douglas, C.J.; Bohlmann, J. Proteome analysis of early somatic embryogenesis in Picea glauca. Proteomics 2005, 5, 461–473. [Google Scholar] [CrossRef]
- Gómez, A.; López, J.A.; Pintos, B.; Camafeita, E.; Bueno, M.Á. Proteomic analysis from haploid and diploid embryos of Quercus suber L. identifies qualitative and quantitative differential expression patterns. Proteomics 2009, 9, 4355–4367. [Google Scholar] [CrossRef]
- Leal, I.; Misra, S. Molecular cloning and characterization of a legumin-like storage protein cDNA of Douglas fir seeds. Plant Mol. Biol. 1993, 21, 709–715. [Google Scholar] [CrossRef]
- Akishev, Z.; Taipakova, S.; Joldybayeva, B.; Zutterling, C.; Smekenov, I.; Ishchenko, A.A.; Zharkov, D.O.; Bissenbaev, A.K.; Saparbaev, M. The major Arabidopsis thaliana apurinic/apyrimidinic endonuclease, ARP is involved in the plant nucleotide incision repair pathway. DNA Repair 2016, 48, 30–42. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teyssier, C.; Rogier, O.; Claverol, S.; Gautier, F.; Lelu-Walter, M.-A.; Duruflé, H. Comprehensive Organ-Specific Profiling of Douglas Fir (Pseudotsuga menziesii) Proteome. Biomolecules 2023, 13, 1400. https://doi.org/10.3390/biom13091400
Teyssier C, Rogier O, Claverol S, Gautier F, Lelu-Walter M-A, Duruflé H. Comprehensive Organ-Specific Profiling of Douglas Fir (Pseudotsuga menziesii) Proteome. Biomolecules. 2023; 13(9):1400. https://doi.org/10.3390/biom13091400
Chicago/Turabian StyleTeyssier, Caroline, Odile Rogier, Stéphane Claverol, Florian Gautier, Marie-Anne Lelu-Walter, and Harold Duruflé. 2023. "Comprehensive Organ-Specific Profiling of Douglas Fir (Pseudotsuga menziesii) Proteome" Biomolecules 13, no. 9: 1400. https://doi.org/10.3390/biom13091400
APA StyleTeyssier, C., Rogier, O., Claverol, S., Gautier, F., Lelu-Walter, M.-A., & Duruflé, H. (2023). Comprehensive Organ-Specific Profiling of Douglas Fir (Pseudotsuga menziesii) Proteome. Biomolecules, 13(9), 1400. https://doi.org/10.3390/biom13091400