
The 2.6 Å Structure of a Tulane Virus Variant with Minor Mutations Leading to Receptor Change

 , ,
, ,
Abstract
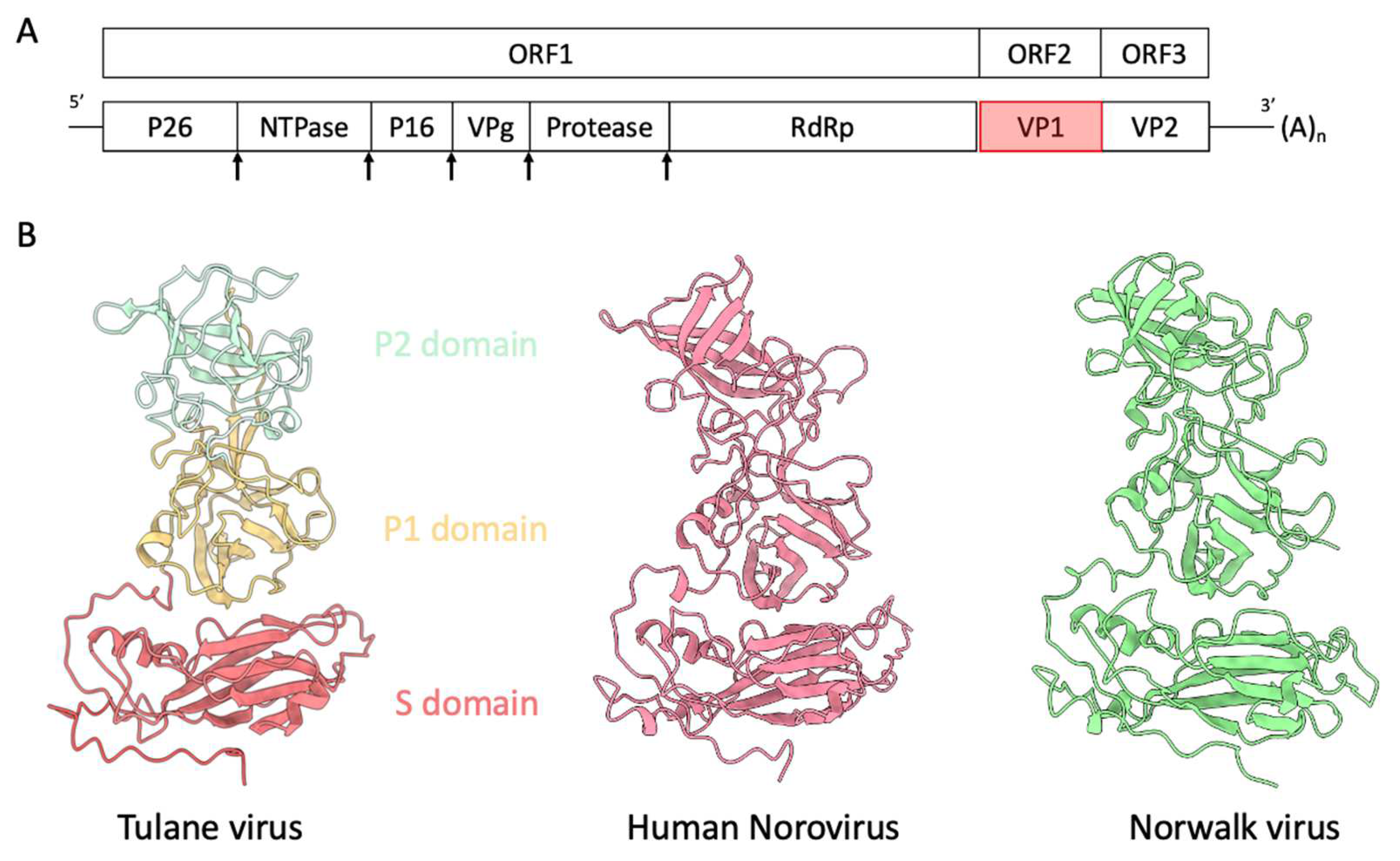
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Purification of Tulane Virus
2.2. Viral RNA Extraction and Genome Sequencing
2.3. Saliva-Based ELISA for Measurement of HBGA Binding
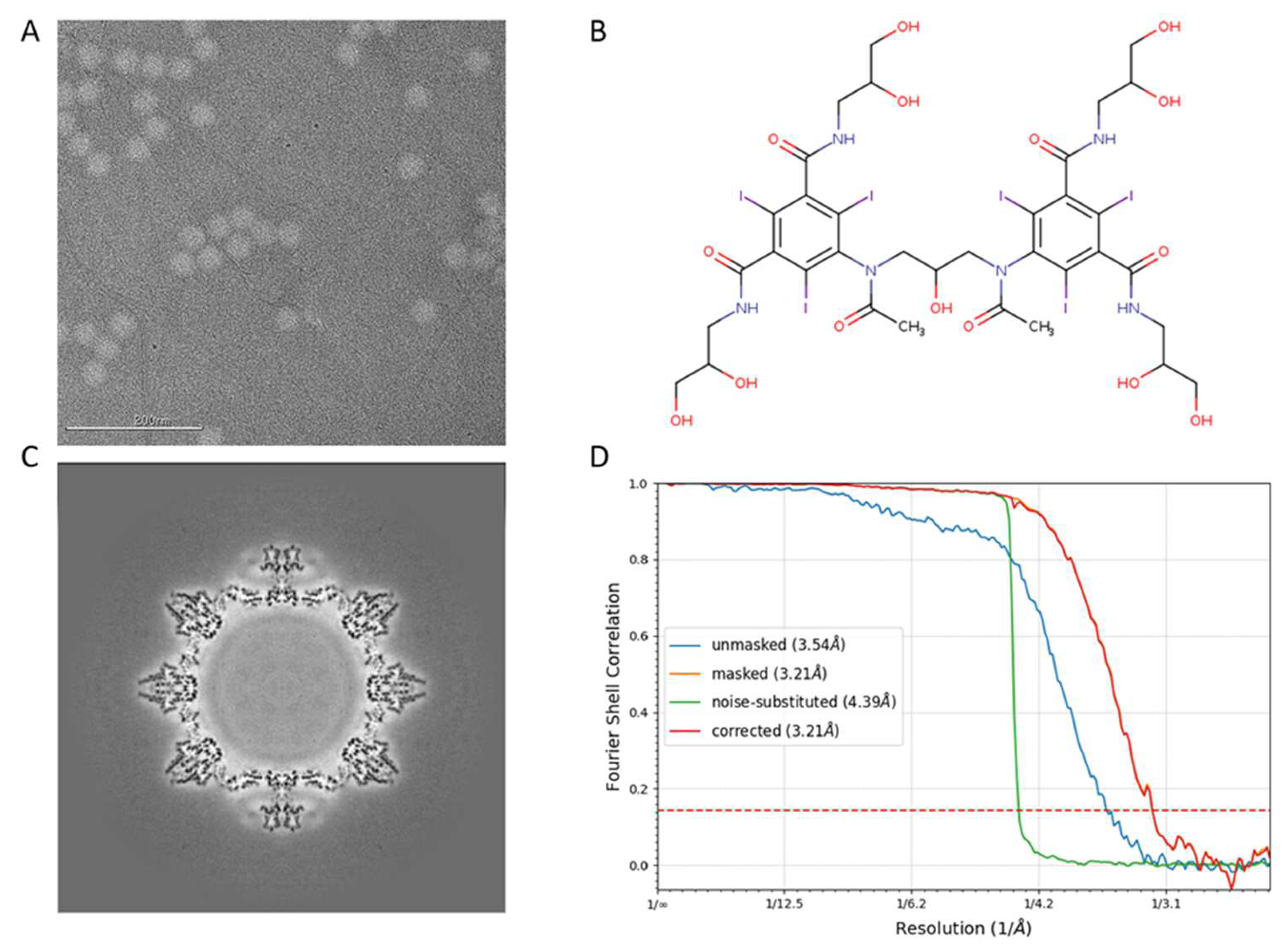
2.4. Cryo-EM Sample Grid Preparation and Data Acquisition
2.5. Image Processing
2.6. Model Refinement
3. Results
3.1. A New TV Variant Has Lost Its Ability to Bind to the Type B HBGA Receptor
3.2. Sequence Analysis of the 9-6-17 TV Strain
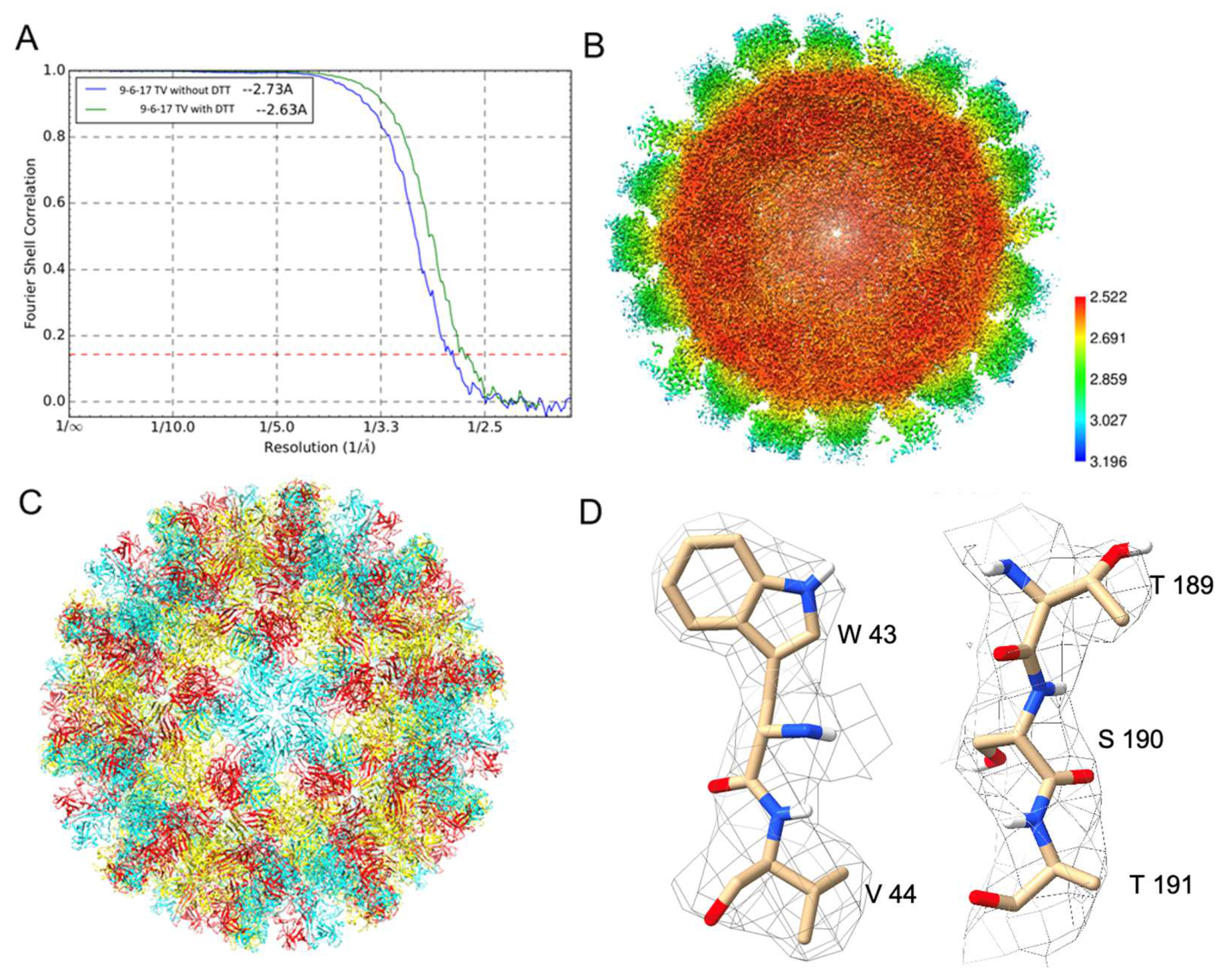
3.3. Structure Determination of the 9-6-17 TV
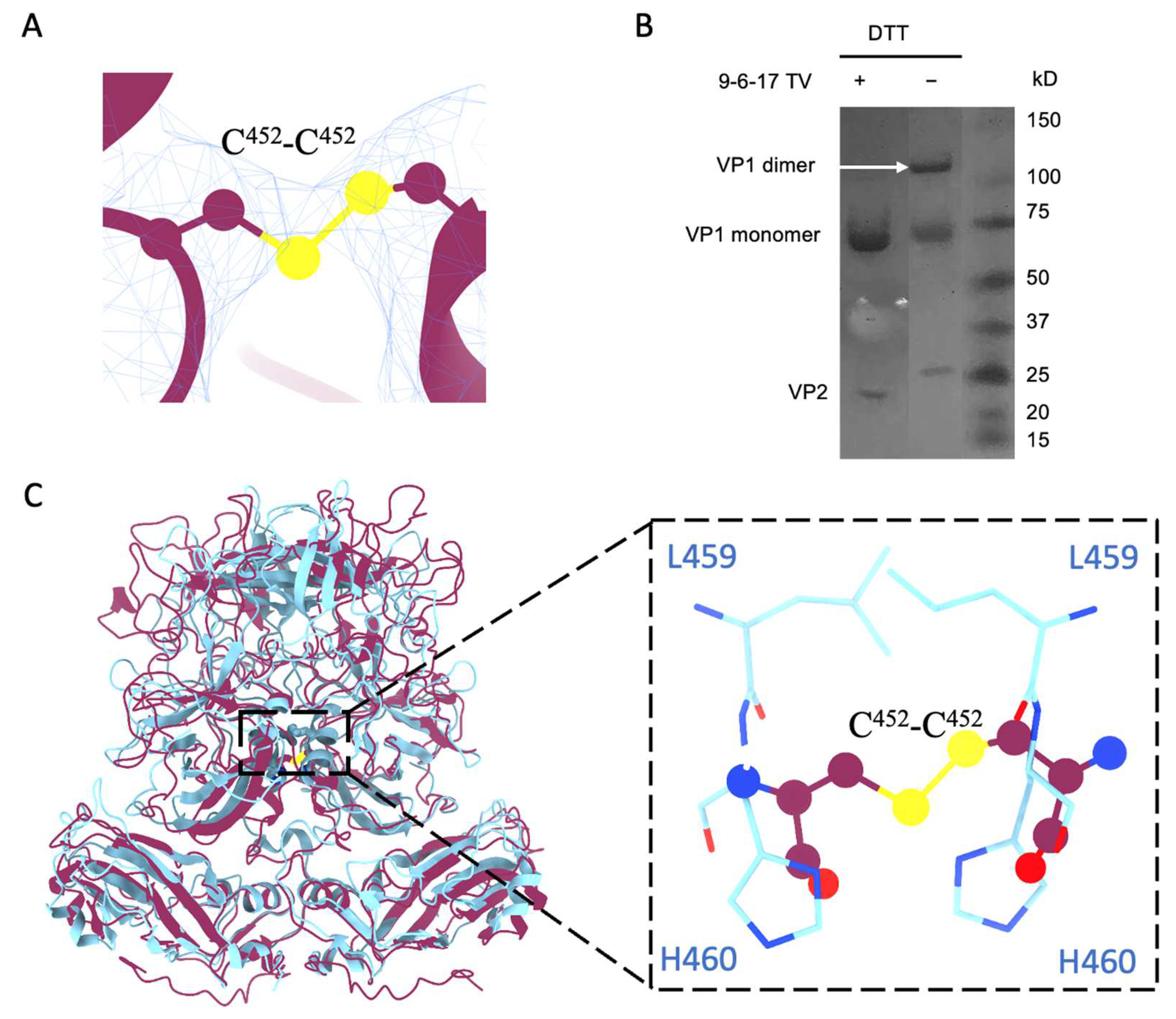
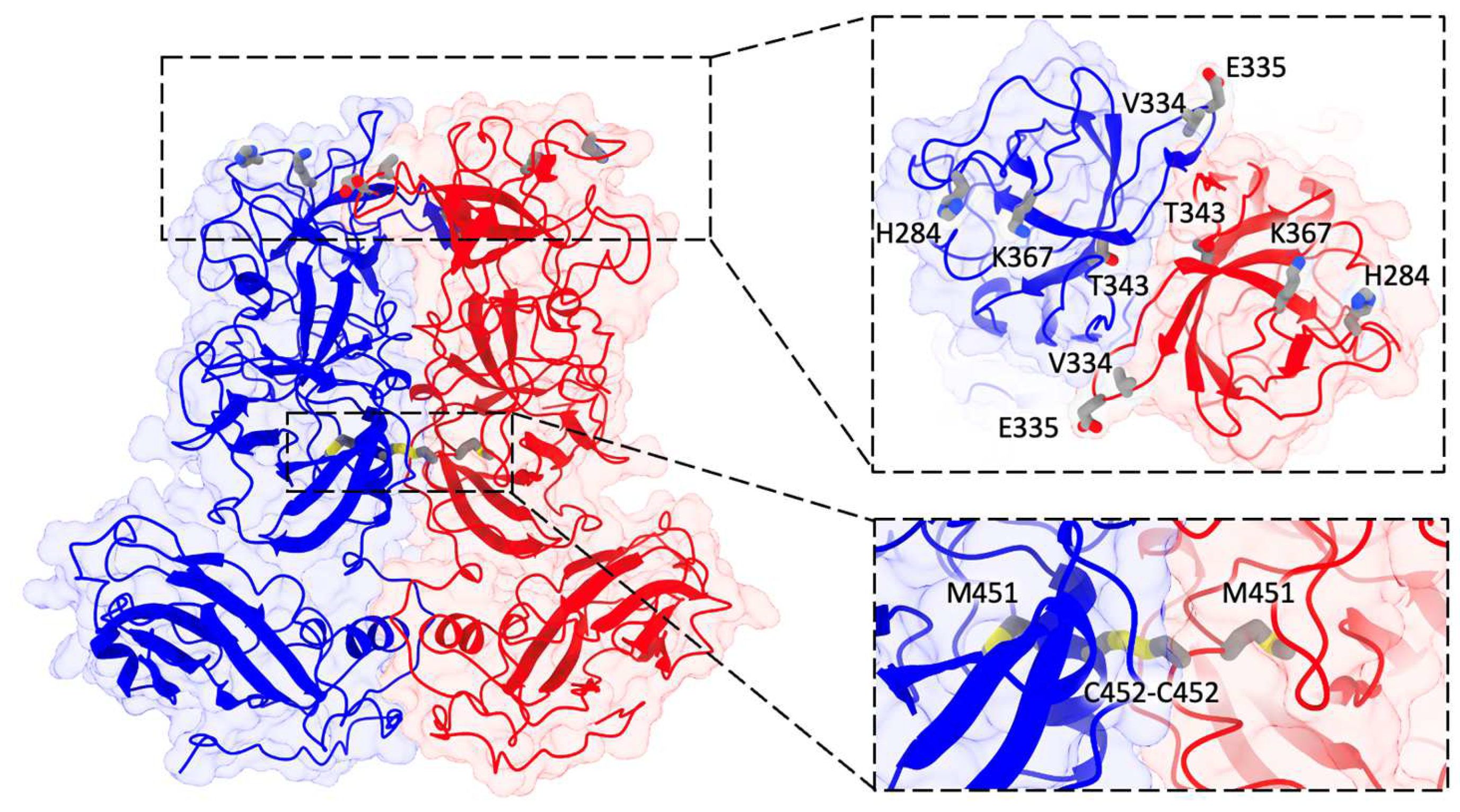
3.4. Disulfide Bond from the R452C Mutation Stabilizes Dimer Interaction
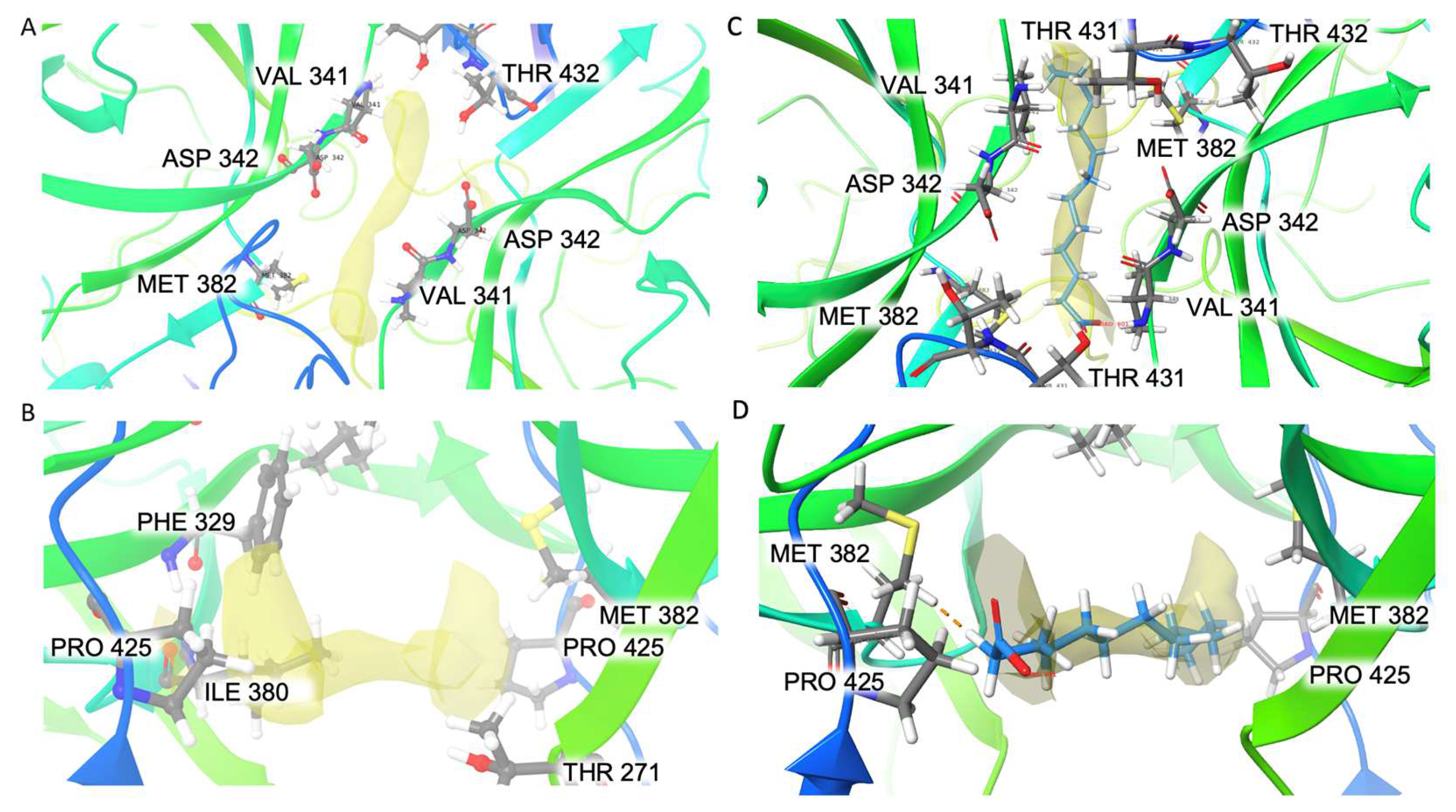
3.5. Elongated Extra Density in the Hydrophobic Pocket in the P Dimer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.A.; et al. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; Tully, D.C.; Mans, J.; Niendorf, S.; Barclay, L.; Cannon, J.L.; Montmayeur, A.M.; Pan, C.Y.; Page, N.; Williams, R.; et al. Emergence of Novel Norovirus GII.4 Variant. Emerg. Infect. Dis. 2024, 30, 163–167. [Google Scholar] [CrossRef]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Farkas, T.; Sestak, K.; Wei, C.; Jiang, X. Characterization of a rhesus monkey calicivirus representing a new genus of Caliciviridae. J. Virol. 2008, 82, 5408–5416. [Google Scholar] [CrossRef]
- Conley, M.J.; McElwee, M.; Azmi, L.; Gabrielsen, M.; Byron, O.; Goodfellow, I.G.; Bhella, D. Calicivirus VP2 forms a portal-like assembly following receptor engagement. Nature 2019, 565, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Wei, C.; Huang, P.; Fan, Q.; Quigley, C.; Xia, M.; Fang, H.; Zhang, X.; Zhong, W.; Klassen, J.S.; et al. Tulane virus recognizes sialic acids as cellular receptors. Sci. Rep. 2015, 5, 11784. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Farkas, T.; Sestak, K.; Jiang, X. Recovery of infectious virus by transfection of in vitro-generated RNA from tulane calicivirus cDNA. J. Virol. 2008, 82, 11429–11436. [Google Scholar] [CrossRef] [PubMed]
- Ravn, V.; Dabelsteen, E. Tissue distribution of histo-blood group antigens. APMIS 2000, 108, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Heggelund, J.E.; Varrot, A.; Imberty, A.; Krengel, U. Histo-blood group antigens as mediators of infections. Curr. Opin. Struct. Biol. 2017, 44, 190–200. [Google Scholar] [CrossRef]
- Yu, G.; Li, K.; Jiang, W. Antibody-based affinity cryo-EM grid. Methods 2016, 100, 16–24. [Google Scholar] [CrossRef]
- Yu, G.; Zhang, D.; Guo, F.; Tan, M.; Jiang, X.; Jiang, W. Cryo-EM structure of a novel calicivirus, Tulane virus. PLoS ONE 2013, 8, e59817. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Grant, T.; Rohou, A.; Grigorieff, N. cisTEM, user-friendly software for single-particle image processing. Elife 2018, 7, e35383. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Guo, F.; Jiang, W. Single particle cryo-electron microscopy and 3-D reconstruction of viruses. Methods Mol. Biol. 2014, 1117, 401–443. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Gonzalez, B.; Vago, F.S.; Jiang, W. High resolution single particle Cryo-EM refinement using JSPR. Prog. Biophys. Mol. Biol. 2021, 160, 37–42. [Google Scholar] [CrossRef]
- Zivanov, J.; Oton, J.; Ke, Z.; von Kugelgen, A.; Pyle, E.; Qu, K.; Morado, D.; Castano-Diez, D.; Zanetti, G.; Bharat, T.A.M.; et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Elife 2022, 11, e83724. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Kimanius, D.; Forsberg, B.O.; Scheres, S.H.; Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 2016, 5, e18722. [Google Scholar] [CrossRef]
- Kimanius, D.; Dong, L.; Sharov, G.; Nakane, T.; Scheres, S.H.W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 2021, 478, 4169–4185. [Google Scholar] [CrossRef]
- Fernandez-Leiro, R.; Scheres, S.H.W. A pipeline approach to single-particle processing in RELION. Acta Crystallogr. D Struct. Biol. 2017, 73, 496–502. [Google Scholar] [CrossRef]
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, R.; Gomez-Blanco, J.; Cuervo, A.; Carazo, J.M.; Sorzano, C.O.S.; Vargas, J. DeepEMhancer: A deep learning solution for cryo-EM volume post-processing. Commun. Biol. 2021, 4, 874. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Song, Y.; Barad, B.A.; Cheng, Y.; Fraser, J.S.; DiMaio, F. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 2016, 5, e17219. [Google Scholar] [CrossRef] [PubMed]
- Tange, O. GNU Parallel—Line Power Tool—Login. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Zhang, D.; Huang, P.; Zou, L.; Lowary, T.L.; Tan, M.; Jiang, X. Tulane virus recognizes the A type 3 and B histo-blood group antigens. J. Virol. 2015, 89, 1419–1427. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Salmen, W.; Chen, R.; Zhou, Y.; Neill, F.; Crowe Jr, J.E.; Prasad, B.V. Atomic structure of the predominant GII.4 human norovirus capsid reveals novel stability and plasticity. Nat. Commun. 2022, 13, 1241. [Google Scholar] [CrossRef]
- Plevka, P.; Perera, R.; Yap, M.L.; Cardosa, J.; Kuhn, R.J.; Rossmann, M.G. Structure of human enterovirus 71 in complex with a capsid-binding inhibitor. Proc. Natl. Acad. Sci. USA 2013, 110, 5463–5467. [Google Scholar] [CrossRef]
- Almand, E.A.; Moore, M.D.; Jaykus, L.A. Norovirus Binding to Ligands Beyond Histo-Blood Group Antigens. Front. Microbiol. 2017, 8, 2549. [Google Scholar] [CrossRef]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.N.; Orchard, R.C.; Kim, A.S.; Stegeman, R.A.; Hsieh, L.L.; Smith, T.J.; Virgin, H.W.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E9201–E9210. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 9-6-17 TV | |
|---|---|
| Sequence length (bp) | 6701 |
| Number of nucleotide mutations in total | 31 |
| Number of nucleotide mutations in ORF2 | 9 |
| Number of nucleotide mutations in ORF3 | 7 |
| Number of amino acid mutations in total | 18 |
| Number of amino acid mutations in VP1 | 8 |
| Number of amino acid mutations in VP2 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Huang, P.; Xu, X.; Vago, F.S.; Li, K.; Klose, T.; Jiang, X.J.; Jiang, W. The 2.6 Å Structure of a Tulane Virus Variant with Minor Mutations Leading to Receptor Change. Biomolecules 2024, 14, 119. https://doi.org/10.3390/biom14010119
Sun C, Huang P, Xu X, Vago FS, Li K, Klose T, Jiang XJ, Jiang W. The 2.6 Å Structure of a Tulane Virus Variant with Minor Mutations Leading to Receptor Change. Biomolecules. 2024; 14(1):119. https://doi.org/10.3390/biom14010119
Chicago/Turabian StyleSun, Chen, Pengwei Huang, Xueyong Xu, Frank S. Vago, Kunpeng Li, Thomas Klose, Xi Jason Jiang, and Wen Jiang. 2024. "The 2.6 Å Structure of a Tulane Virus Variant with Minor Mutations Leading to Receptor Change" Biomolecules 14, no. 1: 119. https://doi.org/10.3390/biom14010119
APA StyleSun, C., Huang, P., Xu, X., Vago, F. S., Li, K., Klose, T., Jiang, X. J., & Jiang, W. (2024). The 2.6 Å Structure of a Tulane Virus Variant with Minor Mutations Leading to Receptor Change. Biomolecules, 14(1), 119. https://doi.org/10.3390/biom14010119