
Mechanistic Insights into Clinically Relevant Ribosome-Targeting Antibiotics


Abstract
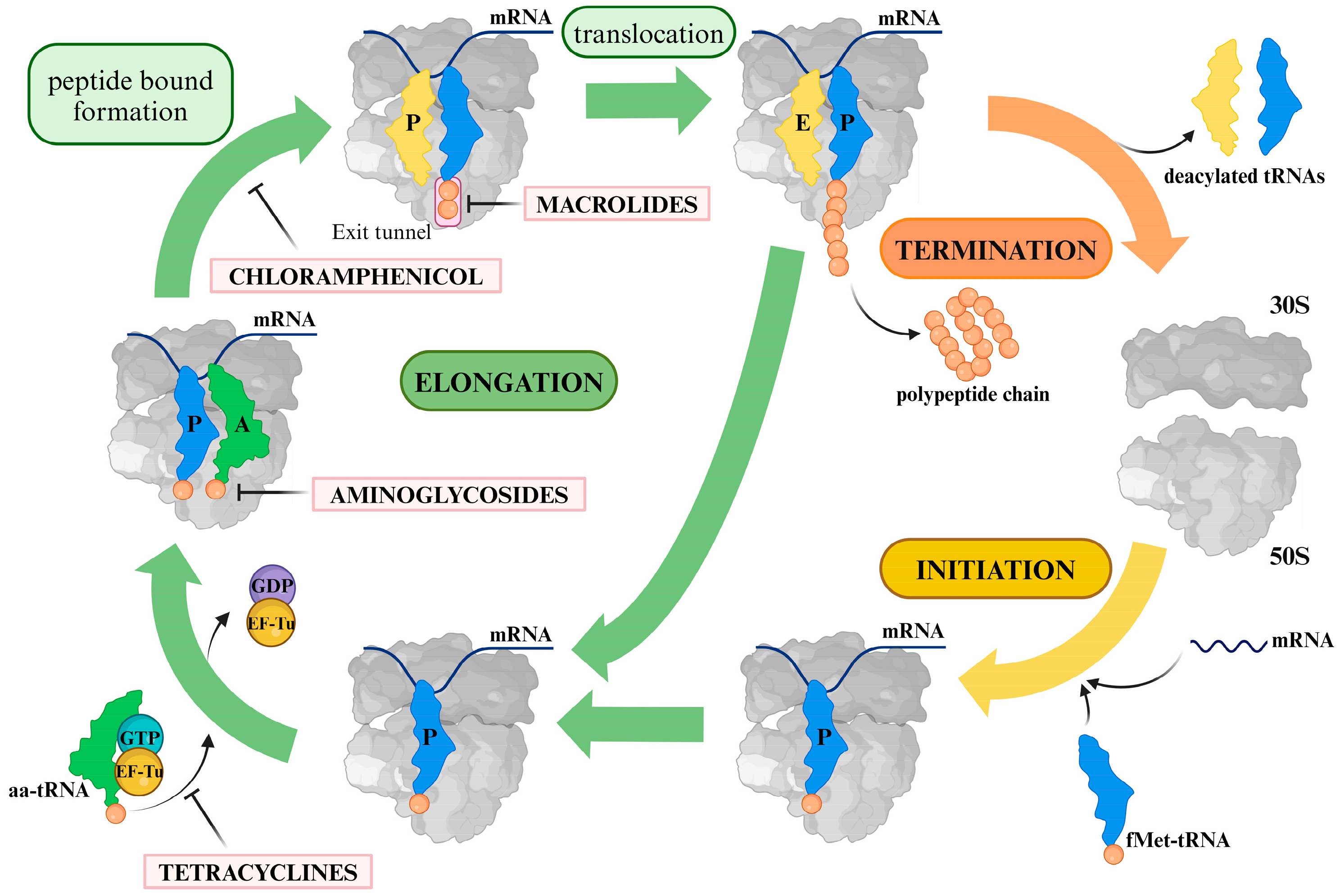
:1. Introduction
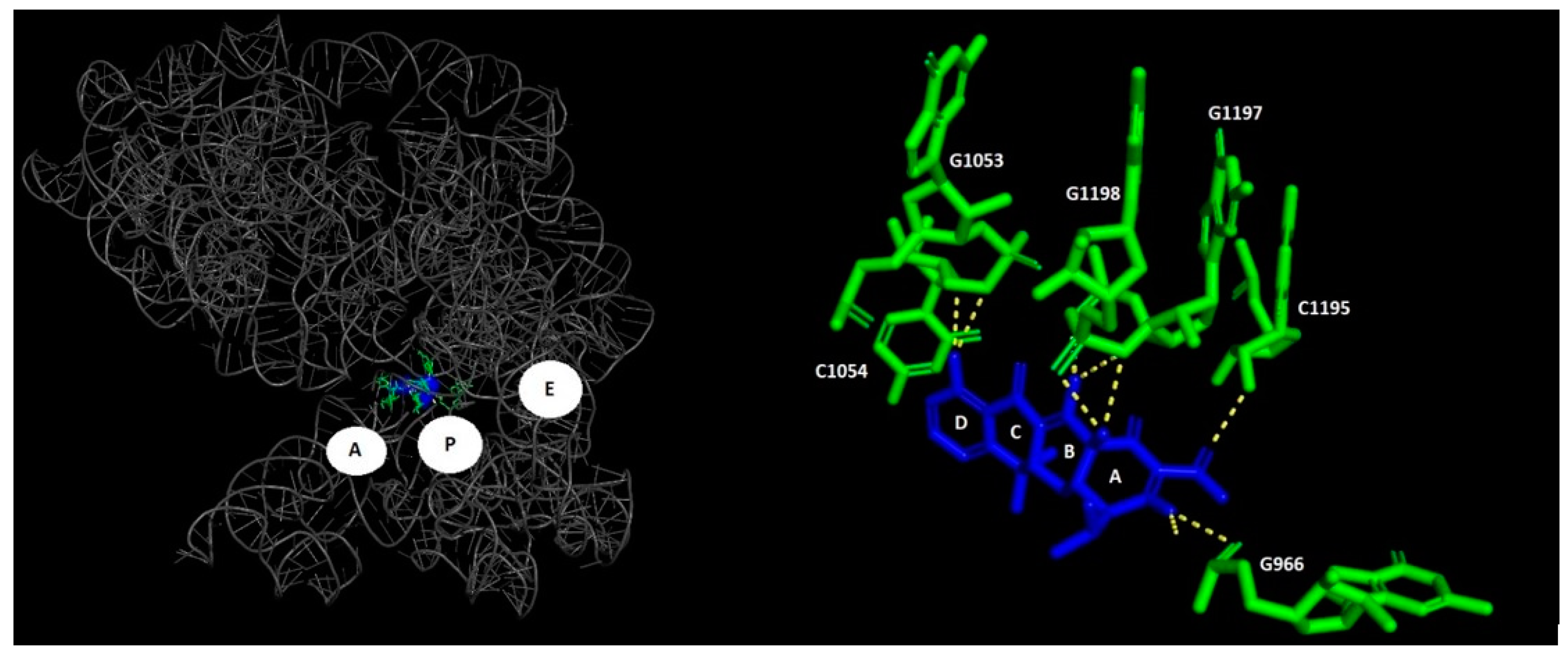
1.1. Macrolides

{kind=link}
{kind=link}
{kind=link}
| Antibiotic | The Primary Reference | Class | Ribosome Binding Site |
|---|---|---|---|
| Erythromycin | [6] | Macrolides | 50S, PTC, exit tunnel |
| Azithromycin | [15] | Macrolides | 50S, PTC, exit tunnel |
| Clarithromycin | [6] | Macrolides | 50S, PTC, exit tunnel |
| Roxithromycin | [6] | Macrolides | 50S, PTC, exit tunnel |
| Telithromycin | [16] | Ketolide | 50S, PTC, exit tunnel |
| Amikacin | [17] | Aminoglycosides | 30S, A-site |
| Gentamicin | [18] | Aminoglycosides | 30S, A-site |
| Tobramycin | [19] | Aminoglycosides | 30S, A-site |
| Netilmicin | [20] | Aminoglycosides | 30S, A-site |
| Neomycin | [18] | Aminoglycosides | 30S, A-site |
| Streptomycin | [21] | Aminoglycosides | 30S, A-site, uS12 |
| Linezolid | [22] | Oxazolidinones | 50S, PTC |
| Clindamycin | [6] | Lincosamides | 50S, PTC |
| Lincomycin | [23] | Lincosamides | 50S, PTC |
| Chloramphenicol | [24] | Amphenicols | 50S, PTC |
| Tetracycline | [25] | Tetracyclines | 30S, A-site |

1.2. Lincosamides
1.3. Oxazolidinones
1.4. Aminoglycosides
1.5. Tetracyclines
1.6. Chloramphenicol
2. General Remarks on the Resistance Mechanisms Associated with Clinically Relevant Antibiotics
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ligon, B.L. Penicillin: Its Discovery and Early Development. Semin. Pediatr. Infect. Dis. 2004, 15, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Cai, Y.; Liu, Y.; An, H.; Deng, K.; Ashraf, M.A.; Zou, L.; Wang, J. Breaking down the Cell Wall: Still an Attractive Antibacterial Strategy. Front. Microbiol. 2022, 13, 952633. [Google Scholar] [CrossRef] [PubMed]
- Poehlsgaard, J.; Douthwaite, S. The Bacterial Ribosome as a Target for Antibiotics. Nat. Rev. Microbiol. 2005, 3, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Mankin, A.S. Macrolide Antibiotics in the Ribosome Exit Tunnel: Species-Specific Binding and Action. Ann. N. Y. Acad. Sci. 2011, 1241, 33–47. [Google Scholar] [CrossRef]
- Vázquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668. [Google Scholar] [CrossRef]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural Basis for the Interaction of Antibiotics with the Peptidyl Transferase Centre in Eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef]
- Garreau De Loubresse, N.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural Basis for the Inhibition of the Eukaryotic Ribosome. Nature 2014 513:7519 2014, 513, 517–522. [Google Scholar] [CrossRef]
- Beckert, B.; Leroy, E.C.; Sothiselvam, S.; Bock, L.V.; Svetlov, M.S.; Graf, M.; Arenz, S.; Abdelshahid, M.; Seip, B.; Grubmüller, H.; et al. Structural and Mechanistic Basis for Translation Inhibition by Macrolide and Ketolide Antibiotics. Nat. Commun. 2021, 12, 4466. [Google Scholar] [CrossRef]
- Mahfouz, A.A.; Said, H.S.; Elfeky, S.M.; Shaaban, M.I. Inhibition of Erythromycin and Erythromycin-Induced Resistance among Staphylococcus Aureus Clinical Isolates. Antibiotics 2023, 12, 503. [Google Scholar] [CrossRef]
- Amsden, G.W. Macrolides versus Azalides: A Drug Interaction Update. Ann. Pharmacother. 1995, 29, 906–917. [Google Scholar] [CrossRef]
- Williams, J.D.; Sefton, A.M. Comparison of Macrolide Antibiotics. J. Antimicrob. Chemother. 1993, 31, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Farag, N.E.; El-Kherbetawy, M.K.; Ismail, H.M.; Abdelrady, A.M.; Toraih, E.A.; Abdelbasset, W.K.; Lashine, R.M.; El-Dosoky, M.; Abed, S.Y.; Ibraheem, K.M.; et al. Differential Effect of Three Macrolide Antibiotics on Cardiac Pathology and Electrophysiology in a Myocardial Infarction Rat Model: Influence on Sodium Nav1.5 Channel Expression. Pharmaceuticals 2021, 14, 597. [Google Scholar] [CrossRef] [PubMed]
- Peeters, T.; Matthijs, G.; Depoortere, I.; Cachet, T.; Hoogmartens, J.; Vantrappen, G. Erythromycin Is a Motilin Receptor Agonist. Am. J. Physiol. 1989, 257, G470–G474. [Google Scholar] [CrossRef]
- Cooper, W.O.; Griffin, M.R.; Arbogast, P.; Hickson, G.B.; Gautam, S.; Ray, W.A. Very Early Exposure to Erythromycin and Infantile Hypertrophic Pyloric Stenosis. Arch. Pediatr. Adolesc. Med. 2002, 156, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Ippolito, J.A.; Ban, N.; Nissen, P.; Moore, P.B.; Steitz, T.A. The Structures of Four Macrolide Antibiotics Bound to the Large Ribosomal Subunit. Mol. Cell. 2002, 10, 117–128. [Google Scholar] [CrossRef]
- Berisio, R.; Harms, J.; Schluenzen, F.; Zarivach, R.; Hansen, H.A.S.; Fucini, P.; Yonath, A. Structural Insight into the Antibiotic Action of Telithromycin against Resistant Mutants. J. Bacteriol. 2003, 185, 4276–4279. [Google Scholar] [CrossRef]
- Seely, S.M.; Parajuli, N.P.; De Tarafder, A.; Ge, X.; Sanyal, S.; Gagnon, M.G. Molecular Basis of the Pleiotropic Effects by the Antibiotic Amikacin on the Ribosome. Nat. Commun. 2023, 14, 4666. [Google Scholar] [CrossRef]
- Borovinskaya, M.A.; Pai, R.D.; Zhang, W.; Schuwirth, B.S.; Holton, J.M.; Hirokawa, G.; Kaji, H.; Kaji, A.; Cate, J.H.D. Structural Basis for Aminoglycoside Inhibition of Bacterial Ribosome Recycling. Nat. Struct. Mol. Biol. 2007, 14, 727–732. [Google Scholar] [CrossRef]
- Yang, G.; Trylska, J.; Tor, Y.; McCammon, J.A. Binding of Aminoglycosidic Antibiotics to the Oligonucleotide A-Site Model and 30S Ribosomal Subunit: Poisson-Boltzmann Model, Thermal Denaturation, and Fluorescence Studies. J. Med. Chem. 2006, 49, 5478–5490. [Google Scholar] [CrossRef]
- Sonousi, A.; Shcherbakov, D.; Vasella, A.; Böttger, E.C.; Crich, D. Synthesis, Ribosomal Selectivity, and Antibacterial Activity of Netilmicin 4′-Derivatives. MedChemComm 2019, 10, 946. [Google Scholar] [CrossRef]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional Insights from the Structure of the 30S Ribosomal Subunit and Its Interactions with Antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N.; Schluenzen, F.; Harms, J.M.; Starosta, A.L.; Connell, S.R.; Fucini, P. The Oxazolidinone Antibiotics Perturb the Ribosomal Peptidyl-Transferase Center and Effect TRNA Positioning. Proc. Natl. Acad. Sci. USA 2008, 105, 13339. [Google Scholar] [CrossRef] [PubMed]
- Matzov, D.; Eyal, Z.; Benhamou, R.I.; Shalev-Benami, M.; Halfon, Y.; Krupkin, M.; Zimmerman, E.; Rozenberg, H.; Bashan, A.; Fridman, M.; et al. Structural Insights of Lincosamides Targeting the Ribosome of Staphylococcus Aureus. Nucleic Acids Res. 2017, 45, 10284–10292. [Google Scholar] [CrossRef]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the Structures of Several Antibiotics Bound to the Bacterial Ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, D.E.; Clemons, W.M.; Carter, A.P.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef]
- Jelić, D.; Antolović, R. From Erythromycin to Azithromycin and New Potential Ribosome-Binding Antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef]
- Ballow, C.H.; Amsden, G.W. Azithromycin: The First Azalide Antibiotic. Ann. Pharmacother. 1992, 26, 1253–1261. [Google Scholar] [CrossRef]
- Ogle, J.M.; Brodersen, D.E.; Clemons, J.; Tarry, M.J.; Carter, A.P.; Ramakrishnan, V. Recognition of Cognate Transfer RNA by the 30S Ribosomal Subunit. Science 2001, 292, 897–902. [Google Scholar] [CrossRef]
- Blondeau, J.M. Immunomodulatory Effects of Macrolides Considering Evidence from Human and Veterinary Medicine. Microorganisms 2022, 10, 2438. [Google Scholar] [CrossRef]
- Langtry, H.D.; Brogdem, R.N. Clarithromycin. A Review of Its Efficacy in the Treatment of Respiratory Tract Infections in Immunocompetent Patients. Drugs 1997, 53, 973–1004. [Google Scholar] [CrossRef]
- Bryskier, A. Novelties in the Field of Macrolides. Expert Opin. Investig. Drugs 1997, 6, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Bryskier, A. Roxithromycin: Review of Its Antimicrobial Activity. J. Antimicrob. Chemother. 1998, 41, 1–21. [Google Scholar] [CrossRef]
- Cooper, B.; Mullins, P.; Jones, M.; Lang, S. Clinical Efficacy of Roxithromycin in the Treatment of Adults with Upper and Lower Respiratory Tract Infection Due to Haemophilus Influenzae: A Meta-Analysis of 12 Clinical Studies. Asian J. Clin. Pediatr. Neonatol. 1994, 2, 299–314. [Google Scholar] [CrossRef]
- Ackermann, G.; Rodloff, A.C. Drugs of the 21st Century: Telithromycin (HMR 3647)—the First Ketolide. J. Antimicrob. Chemother. 2003, 51, 497–511. [Google Scholar] [CrossRef]
- Tu, D.; Blaha, G.; Moore, P.B.; Steitz, T.A. Structures of MLSBK Antibiotics Bound to Mutated Large Ribosomal Subunits Provide a Structural Explanation for Resistance. Cell 2005, 121, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Raja, A.; Lebbos, J.; Kirkpatrick, P. Telithromycin. Nat. Rev. Drug Discov. 2004, 3, 733–734. [Google Scholar] [CrossRef]
- Tenson, T.; Lovmar, M.; Ehrenberg, M. The Mechanism of Action of Macrolides, Lincosamides and Streptogramin B Reveals the Nascent Peptide Exit Path in the Ribosome. J. Mol. Biol. 2003, 330, 1005–1014. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, W.; Yi, J.; Li, B.; Liu, M.; Zhang, M.; Zheng, Y.; Liu, R.; Wu, H.; Zhang, B. Transcriptomics-Guided Investigation of the SLCG_Lrp Regulon Provides New Insights into Its Role for Lincomycin Biosynthesis. Fermentation 2023, 9, 396. [Google Scholar] [CrossRef]
- Leclercq, R. Mechanisms of Resistance to Macrolides and Lincosamides: Nature of the Resistance Elements and Their Clinical Implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef]
- Álvarez, L.A.; Van de Sijpe, G.; Desmet, S.; Metsemakers, W.J.; Spriet, I.; Allegaert, K.; Rozenski, J. Ways to Improve Insights into Clindamycin Pharmacology and Pharmacokinetics Tailored to Practice. Antibiotics 2022, 11, 701. [Google Scholar] [CrossRef]
- Armillei, M.K.; Lomakin, I.B.; Del Rosso, J.Q.; Grada, A.; Bunick, C.G. Scientific Rationale and Clinical Basis for Clindamycin Use in the Treatment of Dermatologic Disease. Antibiotics 2024, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, F.; Zanchi, A.; Stolzuoli, L.; Croci, L.; Cellesi, C. Clindamycin-Resistant Streptococcus Pneumoniae. Emerg. Infect. Dis. 2007, 13, 801. [Google Scholar] [CrossRef] [PubMed]
- Djurkovic-Djakovic, O.; Milenković, V.; Nikolić, A.; Bobić, B.; Grujić, J. Efficacy of Atovaquone Combined with Clindamycin against Murine Infection with a Cystogenic (Me49) Strain of Toxoplasma Gondii. J. Antimicrob. Chemother. 2002, 50, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Foti, C.; Piperno, A.; Scala, A.; Giuffrè, O. Oxazolidinone Antibiotics: Chemical, Biological and Analytical Aspects. Molecules 2021, 26, 4280. [Google Scholar] [CrossRef]
- Bedeni, B.; Chong Gan, W.; Fuh Ng, H.; Fong Ngeow, Y.; Rahman, A.; Sungai Long, J.; Sungai Long, B. Mechanisms of Linezolid Resistance in Mycobacteria. Pharmaceuticals 2023, 16, 784. [Google Scholar] [CrossRef]
- Stefani, S.; Bongiorno, D.; Mongelli, G.; Campanile, F. Linezolid Resistance in Staphylococci. Pharmaceuticals 2010, 3, 1988–2006. [Google Scholar] [CrossRef]
- Behra-Miellet, J.; Calvet, L.; Dubreuil, L. Activity of Linezolid against Anaerobic Bacteria. Int. J. Antimicrob. Agents 2003, 22, 28–34. [Google Scholar] [CrossRef]
- Abou Hassan, O.K.; Karnib, M.; El-Khoury, R.; Nemer, G.; Ahdab-Barmada, M.; BouKhalil, P. Linezolid Toxicity and Mitochondrial Susceptibility: A Novel Neurological Complication in a Lebanese Patient. Front. Pharmacol. 2016, 7, 325. [Google Scholar] [CrossRef]
- Soriano, A.; Miró, O.; Mensa, J. Mitochondrial Toxicity Associated with Linezolid. N. Engl. J. Med. 2005, 353, 2305–2306. [Google Scholar] [CrossRef]
- Dezanet, C.; Kempf, J.; Mingeot-Leclercq, M.P.; Décout, J.L. Amphiphilic Aminoglycosides as Medicinal Agents. Int. J. Mol. Sci. 2020, 21, 7411. [Google Scholar] [CrossRef]
- Davis, B.D. Mechanism of Bactericidal Action of Aminoglycosides. Microbiol. Rev. 1987, 51, 341. [Google Scholar] [CrossRef] [PubMed]
- Allan Drummond, D.; Wilke, C.O. The Evolutionary Consequences of Erroneous Protein Synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Cao, J.; Cheng, A.; Peppelenbosch, M.P.; Pan, Q. Errors in Translational Decoding: TRNA Wobbling or Misincorporation? PLoS Genet. 2019, 15, e1008017. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Chow, C.S. Pseudouridine Modifications Influence Binding of Aminoglycosides to Helix 69 of Bacterial Ribosomes. Org. Biomol. Chem. 2017, 15, 8535. [Google Scholar] [CrossRef]
- Fourmy, D.; Recht, M.I.; Puglisi, J.D. Binding of Neomycin-Class Aminoglycoside Antibiotics to the A-Site of 16 S RRNA. J. Mol. Biol. 1998, 277, 347–362. [Google Scholar] [CrossRef]
- Nicolau, D.P.; Freeman, C.D.; Belliveau, P.P.; Nightingale, C.H.; Ross, J.W.; Quintiliani, R. Experience with a Once-Daily Aminoglycoside Program Administered to 2,184 Adult Patients. Antimicrob. Agents Chemother. 1995, 39, 650. [Google Scholar] [CrossRef]
- Galimand, M.; Sabtcheva, S.; Courvalin, P.; Lambert, T. Worldwide Disseminated ArmA Aminoglycoside Resistance Methylase Gene Is Borne by Composite Transposon Tn1548. Antimicrob. Agents Chemother. 2005, 49, 2949. [Google Scholar] [CrossRef]
- Rather, P.N. Origins of the Aminoglycoside Modifying Enzymes. Drug Resist. Updates 1998, 1, 285–291. [Google Scholar] [CrossRef]
- Heinemann, F.; Leypold, C.; Roman, C.; Schmitt, M.; Schneider, S. X-Ray Crystallography of Tetracycline, Doxycycline and Sancycline. J. Chem. Crystallogr. 2013, 43, 213–222. [Google Scholar] [CrossRef]
- Pioletti, M.; Schlünzen, F.; Harms, J.; Zarivach, R.; Glühmann, M.; Avila, H.; Bashan, A.; Bartels, H.; Auerbach, T.; Jacobi, C.; et al. Crystal Structures of Complexes of the Small Ribosomal Subunit with Tetracycline, Edeine and IF3. EMBO J. 2001, 20, 1829–1839. [Google Scholar] [CrossRef]
- Rusu, A.; Buta, E.L. The Development of Third-Generation Tetracycline Antibiotics and New Perspectives. Pharmaceutics 2021, 13, 2085. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Tetracycline Resistance Determinants: Mechanisms of Action, Regulation of Expression, Genetic Mobility, and Distribution. FEMS Microbiol. Rev. 1996, 19, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ojima-Kato, T.; Nishikawa, Y.; Furukawa, Y.; Kojima, T.; Nakano, H. Nascent MSKIK Peptide Cancels Ribosomal Stalling by Arrest Peptides in Escherichia Coli. J. Biol. Chem. 2023, 299, 104676. [Google Scholar] [CrossRef]
- Chen, C.W.; Pavlova, J.A.; Lukianov, D.A.; Tereshchenkov, A.G.; Makarov, G.I.; Khairullina, Z.Z.; Tashlitsky, V.N.; Paleskava, A.; Konevega, A.L.; Bogdanov, A.A.; et al. Binding and Action of Triphenylphosphonium Analog of Chloramphenicol upon the Bacterial Ribosome. Antibiotics 2021, 10, 390. [Google Scholar] [CrossRef]
- Livermore, D.M. Multiple Mechanisms of Antimicrobial Resistance in Pseudomonas Aeruginosa: Our Worst Nightmare? Clin. Infect. Dis. 2002, 34, 634–640. [Google Scholar] [CrossRef]
- Douthwaite, S.; Hansen, L.H.; Mauvais, P. Macrolide-Ketolide Inhibition of MLS-Resistant Ribosomes Is Improved by Alternative Drug Interaction with Domain II of 23S RRNA. Mol. Microbiol. 2000, 36, 183–193. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef]
- Long, K.S.; Vester, B. Resistance to Linezolid Caused by Modifications at Its Binding Site on the Ribosome. Antimicrob. Agents Chemother. 2012, 56, 603. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Alharby, T.N.; Alanazi, J.; Alanazi, M.; Abdallah, M.H.; Rizvi, S.M.D.; Moin, A.; Khafagy, E.S.; Tabrez, S.; Al Balushi, A.A.; et al. Clinical Resistant Strains of Enterococci and Their Correlation to Reduced Susceptibility to Biocides: Phenotypic and Genotypic Analysis of Macrolides, Lincosamides, and Streptogramins. Antibiotics 2023, 12, 461. [Google Scholar] [CrossRef]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular Basis of Bacterial Resistance to Chloramphenicol and Florfenicol. FEMS Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.H. Tetracycline Antibiotics and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025387. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawczyk, S.J.; Leśniczak-Staszak, M.; Gowin, E.; Szaflarski, W. Mechanistic Insights into Clinically Relevant Ribosome-Targeting Antibiotics. Biomolecules 2024, 14, 1263. https://doi.org/10.3390/biom14101263
Krawczyk SJ, Leśniczak-Staszak M, Gowin E, Szaflarski W. Mechanistic Insights into Clinically Relevant Ribosome-Targeting Antibiotics. Biomolecules. 2024; 14(10):1263. https://doi.org/10.3390/biom14101263
Chicago/Turabian StyleKrawczyk, Szymon J., Marta Leśniczak-Staszak, Ewelina Gowin, and Witold Szaflarski. 2024. "Mechanistic Insights into Clinically Relevant Ribosome-Targeting Antibiotics" Biomolecules 14, no. 10: 1263. https://doi.org/10.3390/biom14101263