
Targeting Protein Aggregation in ALS


Abstract
:
1. Hypothesis of Widespread Aggregation in Proteinopathies
2. Several Proteins Are Aggregated in ALS
3. Therapies Targeting Protein Aggregation in Clinical Trial for ALS
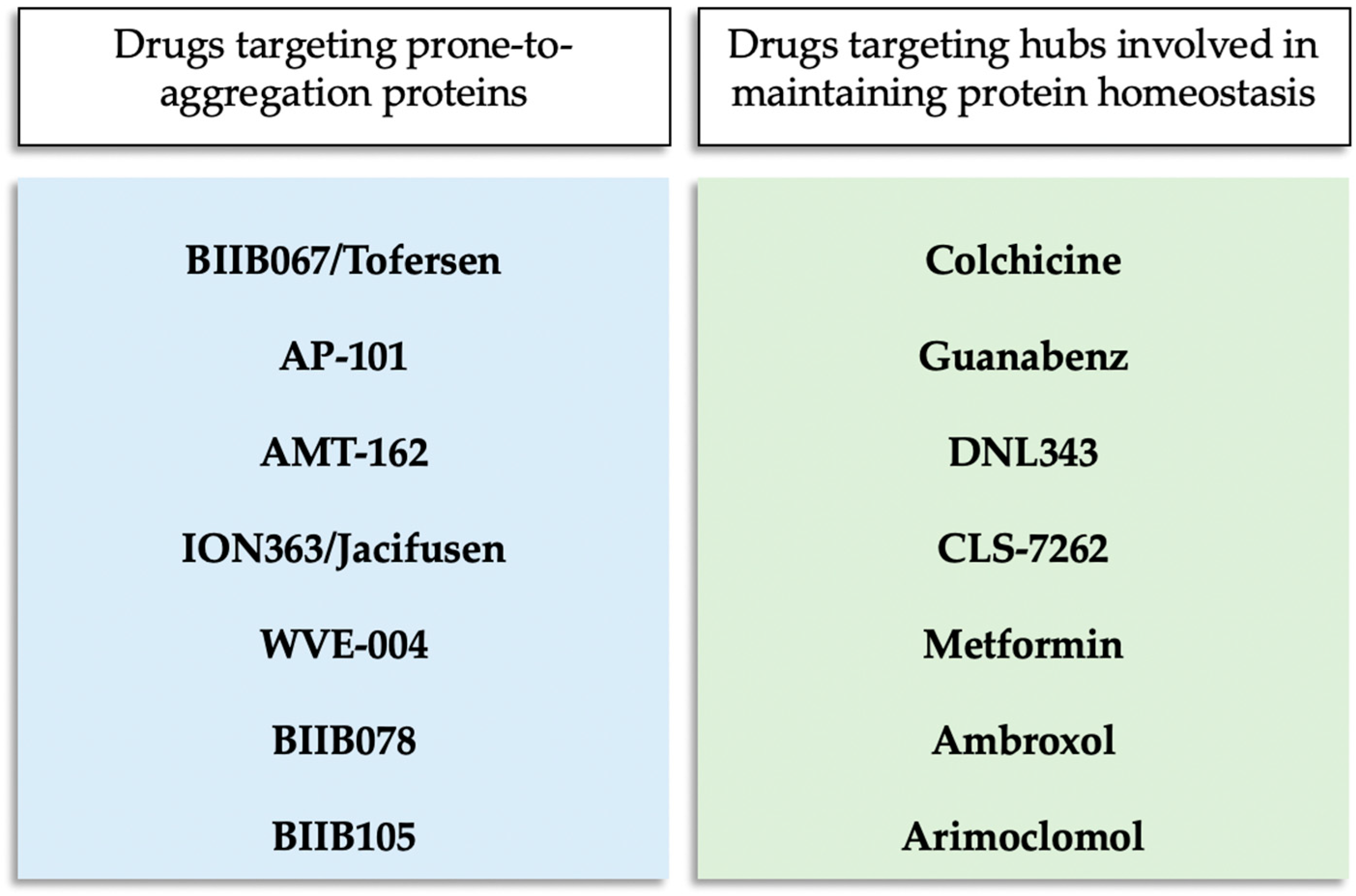
3.1. BIIB067/Tofersen
3.2. AP-101
3.3. AMT-162
3.4. ION363/Jacifusen
3.5. Colchicine
3.6. Guanabenz
3.7. DNL343
3.8. ABBV-CLS-7262
3.9. Metformin
3.10. Ambroxol
3.11. WVE-004
3.12. BIIB078
3.13. BIIB105
3.14. Arimoclomol
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cox, D.; Raeburn, C.; Sui, X.; Hatters, D.M. Protein Aggregation in Cell Biology: An Aggregomics Perspective of Health and Disease. Semin. Cell Dev. Biol. 2020, 99, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Vecchi, G.; Sormanni, P.; Mannini, B.; Vandelli, A.; Tartaglia, G.G.; Dobson, C.M.; Hartl, F.U.; Vendruscolo, M. Proteome-Wide Observation of the Phenomenon of Life on the Edge of Solubility. Proc. Natl. Acad. Sci. USA 2020, 117, 1015–1020. [Google Scholar] [CrossRef]
- Sui, X.; Pires, D.E.V.; Ormsby, A.R.; Cox, D.; Nie, S.; Vecchi, G.; Vendruscolo, M.; Ascher, D.B.; Reid, G.E.; Hatters, D.M. Widespread Remodeling of Proteome Solubility in Response to Different Protein Homeostasis Stresses. Proc. Natl. Acad. Sci. USA 2020, 117, 2422–2431. [Google Scholar] [CrossRef]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C. Elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.-M.; Taylor, J.P.; Parker, R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An Aberrant Phase Transition of Stress Granules Triggered by Misfolded Protein and Prevented by Chaperone Function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Adiutori, R.; Puentes, F.; Bremang, M.; Lombardi, V.; Zubiri, I.; Leoni, E.; Aarum, J.; Sheer, D.; McArthur, S.; Pike, I.; et al. Analysis of Circulating Protein Aggregates as a Route of Investigation into Neurodegenerative Disorders. Brain Commun. 2021, 3, fcab148. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Fani, G.; Bessi, V.; Napolitano, L.; Bagnoli, S.; Ingannato, A.; Neri, L.; Cascella, R.; Matteini, P.; Sorbi, S.; et al. Putative Novel CSF Biomarkers of Alzheimer’s Disease Based on the Novel Concept of Generic Protein Misfolding and Proteotoxicity: The PRAMA Cohort. Transl. Neurodegener. 2024, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Seddighi, S.; Qi, Y.A.; Brown, A.-L.; Wilkins, O.G.; Bereda, C.; Belair, C.; Zhang, Y.-J.; Prudencio, M.; Keuss, M.J.; Khandeshi, A.; et al. Mis-Spliced Transcripts Generate de Novo Proteins in TDP-43-Related ALS/FTD. Sci. Transl. Med. 2024, 16, eadg7162. [Google Scholar] [CrossRef]
- Castro-Gomez, S.; Heneka, M.T. Innate Immune Activation in Neurodegenerative Diseases. Immunity 2024, 57, 790–814. [Google Scholar] [CrossRef]
- Zamiri, K.; Kesari, S.; Paul, K.; Hwang, S.H.; Hammock, B.; Kaczor-Urbanowicz, K.E.; Urbanowicz, A.; Gao, L.; Whitelegge, J.; Fiala, M. Therapy of Autoimmune Inflammation in Sporadic Amyotrophic Lateral Sclerosis: Dimethyl Fumarate and H-151 Downregulate Inflammatory Cytokines in the cGAS-STING Pathway. FASEB J. 2023, 37, e23068. [Google Scholar] [CrossRef]
- Kumar, S.; Thangakani, A.M.; Nagarajan, R.; Singh, S.K.; Velmurugan, D.; Gromiha, M.M. Autoimmune Responses to Soluble Aggregates of Amyloidogenic Proteins Involved in Neurodegenerative Diseases: Overlapping Aggregation Prone and Autoimmunogenic Regions. Sci. Rep. 2016, 6, 22258. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Nery, E.S.M.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Fiala, M.; Lin, J.; Ringman, J.; Kermani-Arab, V.; Tsao, G.; Patel, A.; Lossinsky, A.S.; Graves, M.C.; Gustavson, A.; Sayre, J.; et al. Ineffective Phagocytosis of Amyloid-β by Macrophages of Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2005, 7, 221–232. [Google Scholar] [CrossRef]
- Currais, A.; Fischer, W.; Maher, P.; Schubert, D. Intraneuronal Protein Aggregation as a Trigger for Inflammation and Neurodegeneration in the Aging Brain. FASEB J. 2017, 31, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Bunton-Stasyshyn, R.K.A.; Saccon, R.A.; Fratta, P.; Fisher, E.M.C. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef]
- Wang, L.-Q.; Ma, Y.; Yuan, H.-Y.; Zhao, K.; Zhang, M.-Y.; Wang, Q.; Huang, X.; Xu, W.-C.; Dai, B.; Chen, J.; et al. Cryo-EM Structure of an Amyloid Fibril Formed by Full-Length Human SOD1 Reveals Its Conformational Conversion. Nat. Commun. 2022, 13, 3491. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP Mutations in Amyotrophic Lateral Sclerosis with TDP-43 Neuropathology: A Genetic and Histopathological Analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial Loss of TDP-43 Function Causes Phenotypes of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef]
- Arseni, D.; Chen, R.; Murzin, A.G.; Peak-Chew, S.Y.; Garringer, H.J.; Newell, K.L.; Kametani, F.; Robinson, A.C.; Vidal, R.; Ghetti, B.; et al. TDP-43 Forms Amyloid Filaments with a Distinct Fold in Type A FTLD-TDP. Nature 2023, 620, 898–903. [Google Scholar] [CrossRef]
- Arseni, D.; Hasegawa, M.; Murzin, A.G.; Kametani, F.; Arai, M.; Yoshida, M.; Ryskeldi-Falcon, B. Structure of Pathological TDP-43 Filaments from ALS with FTLD. Nature 2022, 601, 139–143. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging Roles in RNA Processing and Neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef]
- Ikenaka, K.; Ishigaki, S.; Iguchi, Y.; Kawai, K.; Fujioka, Y.; Yokoi, S.; Abdelhamid, R.F.; Nagano, S.; Mochizuki, H.; Katsuno, M.; et al. Characteristic Features of FUS Inclusions in Spinal Motor Neurons of Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2020, 79, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, S.; Hu, J.; Tao, Y.; Xia, W.; Gu, J.; Li, Y.; Cao, Q.; Li, D.; Liu, C. Molecular Structure of an Amyloid Fibril Formed by FUS Low-Complexity Domain. iScience 2022, 25, 103701. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Beck, J.; Poulter, M.; Hensman, D.; Rohrer, J.D.; Mahoney, C.J.; Adamson, G.; Campbell, T.; Uphill, J.; Borg, A.; Fratta, P.; et al. Large C9orf72 Hexanucleotide Repeat Expansions Are Seen in Multiple Neurodegenerative Syndromes and Are More Frequent than Expected in the UK Population. Am. J. Hum. Genet. 2013, 92, 345–353. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Petrauskas, A.; Fortunati, D.L.; Kandi, A.R.; Pothapragada, S.S.; Agrawal, K.; Singh, A.; Huelsmeier, J.; Hillebrand, J.; Brown, G.; Chaturvedi, D.; et al. Structured and Disordered Regions of Ataxin-2 Contribute Differently to the Specificity and Efficiency of mRNP Granule Formation. PLoS Genet. 2024, 20, e1011251. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress Granules as Crucibles of ALS Pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic Reduction of Ataxin-2 Extends Lifespan and Reduces Pathology in TDP-43 Mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Genge, A.; Berube-Desrosiers, M.; Zinman, L.; Shoesmith, C.; Salzman, M.; Schott, R.; Clermont, P.; Bingham, J.; Bowden, E. A Phase 1, Multicenter, Open Label, Single-Ascending Dose Study to Evaluate Safety, Tolerability, and Pharmacokinetics of AP-101 in Familial and Sporadic Amyotrophic Lateral Sclerosis (ALS) (4484). Neurology 2021, 96, 4484. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense Oligonucleotide Silencing of FUS Expression as a Therapeutic Approach in Amyotrophic Lateral Sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef]
- Mandrioli, J.; Crippa, V.; Cereda, C.; Bonetto, V.; Zucchi, E.; Gessani, A.; Ceroni, M.; Chio, A.; D’Amico, R.; Monsurrò, M.R.; et al. Proteostasis and ALS: Protocol for a Phase II, Randomised, Double-Blind, Placebo-Controlled, Multicentre Clinical Trial for Colchicine in ALS (Co-ALS). BMJ Open 2019, 9, e028486. [Google Scholar] [CrossRef]
- Gianferrari, G.; Costantini, R.C.; Crippa, V.; Carra, S.; Bonetto, V.; Pansarasa, O.; Cereda, C.; Zucchi, E.; Martinelli, I.; Simonini, C.; et al. Colchicine Treatment in Amyotrophic Lateral Sclerosis: Safety, Biological and Clinical Effects in a Randomized Clinical Trial. Brain Commun. 2024, 6, fcae304. [Google Scholar] [CrossRef]
- Bella, E.D.; Bersano, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Filosto, M.; Giannini, F.; et al. The Unfolded Protein Response in Amyotrophic Later Sclerosis: Results of a Phase 2 Trial. Brain 2021, 144, 2635–2647. [Google Scholar] [CrossRef]
- Dalla Bella, E.; Tramacere, I.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Eleopra, R.; Filosto, M.; et al. Protein Misfolding, Amyotrophic Lateral Sclerosis and Guanabenz: Protocol for a Phase II RCT with Futility Design (ProMISe Trial). BMJ Open 2017, 7, e015434. [Google Scholar] [CrossRef]
- Sun, L.; Tsai, R.; Yulyaningsih, E.; Fanok, M.; Vissers, M.; Heuberger, J.; Flores, B.; Huang, F.; Kane, L.; Cohen, I.; et al. The Integrated Stress Response Is Modulated by eIF2B Agonist DNL343: Results From Phase 1 Healthy Subject and Phase 1b ALS Patient Studies. (P8-8.010). Neurology 2023, 100, 3555. [Google Scholar] [CrossRef]
- Yulyaningsih, E.; Suh, J.H.; Fanok, M.H.; Chau, R.; Solanoy, H.; Takahashi, R.; Bakardjiev, A.I.; Becerra, I.; Benitez, N.B.; Chiu, C.-L.; et al. DNL343 Is an Investigational CNS Penetrant eIF2B Activator That Prevents and Reverses the Effects of Neurodegeneration Caused by the Integrated Stress Response. bioRxiv 2024. [Google Scholar] [CrossRef]
- Cho, W.; Jeong, A.; Malik, P.; Boiser, J.; Huang, X.; Rosebraugh, M. A Phase 1 First-in-Human Study to Investigate the Safety, Tolerability and Food Effect of ABBV-CLS-7262 (P6-4.002). Neurology 2023, 100, 4188. [Google Scholar] [CrossRef]
- Van Den Berg, L.H.; Rothstein, J.D.; Shaw, P.J.; Babu, S.; Benatar, M.; Bucelli, R.C.; Genge, A.; Glass, J.D.; Hardiman, O.; Libri, V.; et al. Safety, Tolerability, and Pharmacokinetics of Antisense Oligonucleotide BIIB078 in Adults with C9orf72-Associated Amyotrophic Lateral Sclerosis: A Phase 1, Randomised, Double Blinded, Placebo-Controlled, Multiple Ascending Dose Study. Lancet Neurol. 2024, 23, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Hansen, T.; Rom, D.; Geist, M.A.; Blaettler, T.; Camu, W.; Kuzma-Kozakiewicz, M.; Van Den Berg, L.H.; Morales, R.J.; Chio, A.; et al. Safety and Efficacy of Arimoclomol in Patients with Early Amyotrophic Lateral Sclerosis (ORARIALS-01): A Randomised, Double-Blind, Placebo-Controlled, Multicentre, Phase 3 Trial. Lancet Neurol. 2024, 23, 687–699. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022, 19, 1248–1258. [Google Scholar] [CrossRef]
- Buratti, E. Targeting TDP-43 Proteinopathy with Drugs and Drug-like Small Molecules. Br. J. Pharmacol. 2021, 178, 1298–1315. [Google Scholar] [CrossRef]
- Crippa, V.; Cicardi, M.E.; Ramesh, N.; Seguin, S.J.; Ganassi, M.; Bigi, I.; Diacci, C.; Zelotti, E.; Baratashvili, M.; Gregory, J.M.; et al. The Chaperone HSPB8 Reduces the Accumulation of Truncated TDP-43 Species in Cells and Protects against TDP-43-Mediated Toxicity. Hum. Mol. Genet. 2016, 25, 3908–3924. [Google Scholar] [CrossRef]
- Crippa, V.; D’Agostino, V.G.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Loffredo, R.; Pancher, M.; Piccolella, M.; Galbiati, M.; et al. Transcriptional Induction of the Heat Shock Protein B8 Mediates the Clearance of Misfolded Proteins Responsible for Motor Neuron Diseases. Sci. Rep. 2016, 6, 22827. [Google Scholar] [CrossRef]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein Quality Control of RNP Granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef]
- Holmes, B.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Guanabenz: A Review of Its Pharmacodynamic Properties and Therapeutic Efficacy in Hypertension. Drugs 1983, 26, 212–229. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef] [PubMed]
- Van Der Knaap, M.S.; Pronk, J.C.; Scheper, G.C. Vanishing White Matter Disease. Lancet Neurol. 2006, 5, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Mallucci, G.R. The Unfolded Protein Response: Mechanisms and Therapy of Neurodegeneration. Brain 2016, 139, 2113–2121. [Google Scholar] [CrossRef]
- Craig, R.A.; De Vicente, J.; Estrada, A.A.; Feng, J.A.; Lexa, K.W.; Canet, M.J.; Dowdle, W.E.; Erickson, R.I.; Flores, B.N.; Haddick, P.C.G.; et al. Discovery of DNL343: A Potent, Selective, and Brain-Penetrant eIF2B Activator Designed for the Treatment of Neurodegenerative Diseases. J. Med. Chem. 2024, 67, 5758–5782. [Google Scholar] [CrossRef]
- Marlin, E.; Viu-Idocin, C.; Arrasate, M.; Aragón, T. The Role and Therapeutic Potential of the Integrated Stress Response in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 7823. [Google Scholar] [CrossRef]
- Bugallo, R.; Marlin, E.; Baltanás, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragón, T. Fine Tuning of the Unfolded Protein Response by ISRIB Improves Neuronal Survival in a Model of Amyotrophic Lateral Sclerosis. Cell Death Dis. 2020, 11, 397. [Google Scholar] [CrossRef]
- Zu, T.; Guo, S.; Bardhi, O.; Ryskamp, D.A.; Li, J.; Tusi, S.K.; Engelbrecht, A.; Klippel, K.; Chakrabarty, P.; Nguyen, L.; et al. Metformin Inhibits RAN Translation through PKR Pathway and Mitigates Disease in C9orf72 ALS/FTD Mice. Proc. Natl. Acad. Sci. USA 2020, 117, 18591–18599. [Google Scholar] [CrossRef]
- Blonde, L.; Dipp, S.; Cadena, D. Combination Glucose-Lowering Therapy Plans in T2DM: Case-Based Considerations. Adv. Ther. 2018, 35, 939–965. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG-Initiated Translation Directed by Microsatellite Expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Malerba, M.; Ragnoli, B. Ambroxol in the 21st Century: Pharmacological and Clinical Update. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1119–1129. [Google Scholar] [CrossRef]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Ron, I.; Horowitz, M. ER Retention and Degradation as the Molecular Basis Underlying Gaucher Disease Heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic Reticulum Stress Is Important for the Manifestations of α-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Bouscary, A.; Quessada, C.; Mosbach, A.; Callizot, N.; Spedding, M.; Loeffler, J.-P.; Henriques, A. Ambroxol Hydrochloride Improves Motor Functions and Extends Survival in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2019, 10, 883. [Google Scholar] [CrossRef]
- Liu, Y.; Andreucci, A.; Iwamoto, N.; Yin, Y.; Yang, H.; Liu, F.; Bulychev, A.; Hu, X.S.; Lin, X.; Lamore, S.; et al. Preclinical Evaluation of WVE-004, Aninvestigational Stereopure Oligonucleotide Forthe Treatment of C9orf72-Associated ALS or FTD. Mol. Ther. Nucleic Acids 2022, 28, 558–570. [Google Scholar] [CrossRef]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.M.G.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9ORF72 Repeat Expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Keon, M.; Musrie, B.; Dinger, M.; Brennan, S.E.; Santos, J.; Saksena, N.K. Destination Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 596006. [Google Scholar] [CrossRef]
- Jack, C.R.; Therneau, T.M.; Weigand, S.D.; Wiste, H.J.; Knopman, D.S.; Vemuri, P.; Lowe, V.J.; Mielke, M.M.; Roberts, R.O.; Machulda, M.M.; et al. Prevalence of Biologically vs Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging—Alzheimer’s Association Research Framework. JAMA Neurol. 2019, 76, 1174–1183. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug Development in Alzheimer’s Disease: The Path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Morimoto, S. iPSC-Based Disease Modeling and Drug Discovery in Cardinal Neurodegenerative Disorders. Cell Stem Cell 2022, 29, 189–208. [Google Scholar] [CrossRef] [PubMed]
- 117TH CONGRESS 2D SESSION. S.5002 Bill–FDA Modernization Act 2.0. 2022, 1–3. Available online: https://www.congress.gov/bill/117th-congress/senate-bill/5002 (accessed on 22 August 2024).
- Zushin, P.-J.H.; Mukherjee, S.; Wu, J.C. FDA Modernization Act 2.0: Transitioning beyond Animal Models with Human Cells, Organoids, and AI/ML-Based Approaches. J. Clin. Investig. 2023, 133, e175824. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Name | Type | Target | Sponsor | Clinical Trial | References |
|---|---|---|---|---|---|
| BIIB067/Tofersen | ASO | SOD1 | Biogen | NCT02623699; NCT03070119 | [41,42] |
| AP-101 | antibody | SOD1 | AL-S Pharma AG | NCT05039099 | [43] |
| AMT-162 | miRNA | SOD1 | UniQure Biopharma BV | NCT06100276 | - |
| ION363/Jacifusen | ASO | FUS | Ionis Pharmaceuticals | NCT04768972 | [44] |
| Colchicine | Small molecule | HSPB8 | Azienda Ospedaliero-Universitaria di Modena | NCT03693781 | [45,46] |
| Guanabenz | Small molecule | PPP1R15A | Fondazione IRCCS Istituto Neurologico Carlo Besta | EudraCT 2017-001042-10 | [47,48] |
| DNL343 | Small molecule | eIF2B | Denali Therapeutics/ Massachusetts General Hospital | NCT05006352; NCT05842941 | [49,50] |
| CLS-7262 | Small molecule | eIF2B | AbbVie, Calico Life Sciences/Massachusetts General Hospital | NCT05740813 | [51] |
| Metformin | Small molecule | PKR pathway | University of Florida | NCT04220021 | - |
| Ambroxol | Small molecule | GBA2 | The Florey Institute of Neuroscience and Mental Health | NCT05959850 | - |
| Name | Type | Target | Sponsor | Clinical Trial | References |
|---|---|---|---|---|---|
| WVE-004 | ASO | C9orf72 | Wave Life Sciences | NCT04931862 | - |
| BIIB078 | ASO | C9orf72 | Biogen | NCT03626012 | [52] |
| BIIB105 | ASO | ataxin-2 | Biogen | NCT04494256 | - |
| Arimoclomol | Small molecule | HSP70 | Orphazyme | NCT03491462 | [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perni, M.; Mannini, B. Targeting Protein Aggregation in ALS. Biomolecules 2024, 14, 1324. https://doi.org/10.3390/biom14101324
Perni M, Mannini B. Targeting Protein Aggregation in ALS. Biomolecules. 2024; 14(10):1324. https://doi.org/10.3390/biom14101324
Chicago/Turabian StylePerni, Michele, and Benedetta Mannini. 2024. "Targeting Protein Aggregation in ALS" Biomolecules 14, no. 10: 1324. https://doi.org/10.3390/biom14101324
APA StylePerni, M., & Mannini, B. (2024). Targeting Protein Aggregation in ALS. Biomolecules, 14(10), 1324. https://doi.org/10.3390/biom14101324