



Exposure to Metals, Pesticides, and Air Pollutants: Focus on Resulting DNA Methylation Changes in Neurodegenerative Diseases



Abstract
:1. Introduction
2. Metals, Pesticides, and Air Pollution as Risk Factors for Age-Related Neurodegenerative Diseases
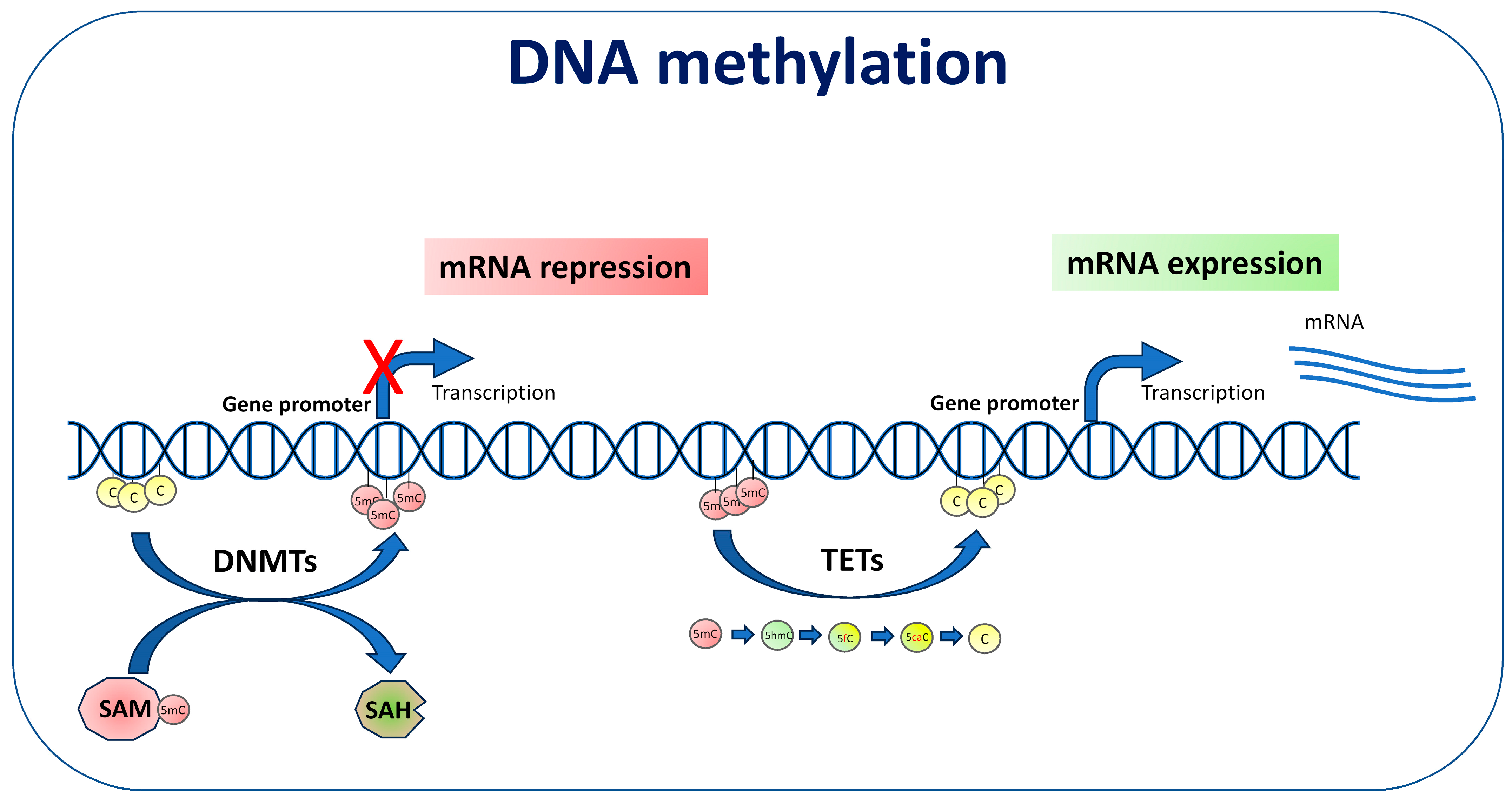
3. DNA Methylation
4. DNA Methylation and Neurodegeneration
5. DNA Methylation Changes Induced by Metals, Pesticides, and Air Pollutants in Neurodegenerative Diseases
5.1. In Vitro and In Vivo Studies
5.2. Human Studies
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Risk Reduction of Cognitive Decline and Dementia: WHO Guidelines; World Health Organization: Geneva, Switzerland, 2019; Available online: https://www.ncbi.nlm.nih.gov/books/NBK542796/ (accessed on 15 October 2024).
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.G.; Andrew, A.S.; Traynor, B.J.; Chiò, A.; Butt, T.H.; Stommel, E.W. Gene-Environment-Time Interactions in Neurodegenerative Diseases: Hypotheses and Research Approaches. Ann. Neurosci. 2018, 25, 261–267. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Nabi, M.; Tabassum, N. Role of Environmental Toxicants on Neurodegenerative Disorders. Front. Toxicol. 2022, 4, 837579. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Cookson, M.R.; Van Den Bosch, L.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Malik, R.; Wiedau, M. Therapeutic Approaches Targeting Protein Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Renton, A.E.; Fulton-Howard, B.; Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. The complex genetic architecture of Alzheimer’s disease: Novel insights and future directions. eBioMedicine 2023, 90, 104511. [Google Scholar] [CrossRef]
- Ben-Shlomo, Y.; Darweesh, S.; Llibre-Guerra, J.; Marras, C.; San Luciano, M.; Tanner, C. The epidemiology of Parkinson’s disease. Lancet 2024, 403, 283–292. [Google Scholar] [CrossRef]
- Rizzuti, M.; Sali, L.; Melzi, V.; Scarcella, S.; Costamagna, G.; Ottoboni, L.; Quetti, L.; Brambilla, L.; Papadimitriou, D.; Verde, F.; et al. Genomic and transcriptomic advances in amyotrophic lateral sclerosis. Ageing Res. Rev. 2023, 92, 102126. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedè, F. Gene-environment interactions in Alzheimer disease: The emerging role of epigenetics. Nat. Rev. Neurol. 2022, 18, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Savelieff, M.G.; Jang, D.G.; Hur, J.; Feldman, E.L. The amyotrophic lateral sclerosis exposome: Recent advances and future directions. Nat. Rev. Neurol. 2023, 19, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Ahmed, M.; Ahmad, S.; Sahoo, C.R.; Iqubal, M.K.; Haque, S.E. Environmental neurotoxic pollutants: Review. Environ. Sci. Pollut. Res. Int. 2020, 27, 41175–41198. [Google Scholar] [CrossRef]
- Kang, Y.J.; Tan, H.Y.; Lee, C.Y.; Cho, H. An Air Particulate Pollutant Induces Neuroinflammation and Neurodegeneration in Human Brain Models. Adv. Sci. 2021, 8, e2101251. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hernández, M.I.; Acosta-Saavedra, L.C.; Hernández-Kelly, L.C.; Loaeza-Loaeza, J.; Ortega, A. Microglial Activation in Metal Neurotoxicity: Impact in Neurodegenerative Diseases. Biomed. Res. Int. 2023, 2023, 7389508. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, M.; Benevenuto, S.G.M.; Tomasini, P.P.; Yariwake, V.Y.; de Oliveira Alves, N.; Rahmeier, F.L.; da Cruz Fernandes, M.; Moura, D.J.; Nascimento Saldiva, P.H.; Veras, M.M. Concentrated ambient fine particulate matter (PM2.5) exposure induce brain damage in pre and postnatal exposed mice. Neurotoxicology 2020, 79, 127–141. [Google Scholar] [CrossRef]
- Shih, C.H.; Chen, J.K.; Kuo, L.W.; Cho, K.H.; Hsiao, T.C.; Lin, Z.W.; Lin, Y.S.; Kang, J.H.; Lo, Y.C.; Chuang, K.J.; et al. Chronic pulmonary exposure to traffic-related fine particulate matter causes brain impairment in adult rats. Part. Fibre Toxicol. 2018, 15, 44. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Vojdani, A.; Blaurock-Busch, E.; Busch, Y.; Friedle, A.; Franco-Lira, M.; Sarathi-Mukherjee, P.; Martínez-Aguirre, X.; Park, S.B.; Torres-Jardón, R.; et al. Air pollution and children: Neural and tight junction antibodies and combustion metals; the role of barrier breakdown and brain immunity in neurodegeneration. J. Alzheimers Dis. 2015, 43, 1039–1058. [Google Scholar] [CrossRef]
- Calderón-Garcidueñas, L.; Herrera-Soto, A.; Jury, N.; Maher, B.A.; González-Maciel, A.; Reynoso-Robles, R.; Ruiz-Rudolph, P.; van Zundert, B.; Varela-Nallar, L. Reduced repressive epigenetic marks, increased DNA damage and Alzheimer’s disease hallmarks in the brain of humans and mice exposed to particulate urban air pollution. Environ. Res. 2020, 183, 109226. [Google Scholar] [CrossRef]
- Cheng, H.; Yang, B.; Ke, T.; Li, S.; Yang, X.; Aschner, M.; Chen, P. Mechanisms of Metal-Induced Mitochondrial Dysfunction in Neurological Disorders. Toxics 2021, 9, 142. [Google Scholar] [CrossRef]
- Bustamante-Barrientos, F.A.; Luque-Campos, N.; Araya, M.J.; Lara-Barba, E.; de Solminihac, J.; Pradenas, C.; Molina, L.; Herrera-Luna, Y.; Utreras-Mendoza, Y.; Elizondo-Vega, R.; et al. Mitochondrial dysfunction in neurodegenerative disorders: Potential therapeutic application of mitochondrial transfer to central nervous system-residing cells. J. Transl. Med. 2023, 21, 613. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.D.; Patisaul, H.B. Environmental Mechanisms of Neurodevelopmental Toxicity. Curr. Environ. Health Rep. 2018, 5, 145–157. [Google Scholar] [CrossRef]
- Modgil, S.; Lahiri, D.K.; Sharma, V.L.; Anand, A. Role of early life exposure and environment on neurodegeneration: Implications on brain disorders. Transl. Neurodegener. 2014, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, L.G.; Bodin, L. Occupational Exposures and Neurodegenerative Diseases-A Systematic Literature Review and Meta-Analyses. Int. J. Environ. Res. Public Health 2019, 16, 337. [Google Scholar] [CrossRef]
- Cariccio, V.L.; Samà, A.; Bramanti, P.; Mazzon, E. Mercury Involvement in Neuronal Damage and in Neurodegenerative Diseases. Biol. Trace Elem. Res. 2019, 187, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef]
- Andrew, A.S.; Chen, C.Y.; Caller, T.A.; Tandan, R.; Henegan, P.L.; Jackson, B.P.; Hall, B.P.; Bradley, W.G.; Stommel, E.W. Toenail mercury Levels are associated with amyotrophic lateral sclerosis risk. Muscle Nerve 2018, 58, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.S.; O’Brien, K.M.; Jackson, B.P.; Sandler, D.P.; Kaye, W.E.; Wagner, L.; Stommel, E.W.; Horton, D.K.; Mehta, P.; Weinberg, C.R. Keratinous biomarker of mercury exposure associated with amyotrophic lateral sclerosis risk in a nationwide U.S. study. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 420–427. [Google Scholar] [CrossRef]
- Thakur, M.; Rachamalla, M.; Niyogi, S.; Datusalia, A.K.; Flora, S.J.S. Molecular Mechanism of Arsenic-Induced Neurotoxicity including Neuronal Dysfunctions. Int. J. Mol. Sci. 2021, 22, 10077. [Google Scholar] [CrossRef]
- Niño, S.A.; Martel-Gallegos, G.; Castro-Zavala, A.; Ortega-Berlanga, B.; Delgado, J.M.; Hernández-Mendoza, H.; Romero-Guzmán, E.; Ríos-Lugo, J.; Rosales-Mendoza, S.; Jiménez-Capdeville, M.E.; et al. Chronic Arsenic Exposure Increases Aβ(1-42) Production and Receptor for Advanced Glycation End Products Expression in Rat Brain. Chem. Res. Toxicol. 2018, 31, 13–21. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Rahman, M.S.; Rashid, M.M.; Kim, B. Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics 2021, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Anyachor, C.P.; Dooka, D.B.; Orish, C.N.; Amadi, C.N.; Bocca, B.; Ruggieri, F.; Senofonte, M.; Frazzoli, C.; Orisakwe, O.E. Mechanistic considerations and biomarkers level in nickel-induced neurodegenerative diseases: An updated systematic review. IBRO Neurosci. Rep. 2022, 13, 136–146. [Google Scholar] [CrossRef]
- Strumylaite, L.; Kregzdyte, R.; Kucikiene, O.; Baranauskiene, D.; Simakauskiene, V.; Naginiene, R.; Damuleviciene, G.; Lesauskaite, V.; Zemaitiene, R. Alzheimer’s Disease Association with Metals and Metalloids Concentration in Blood and Urine. Int. J. Environ. Res. Public Health 2022, 19, 7309. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259. [Google Scholar] [CrossRef]
- Martins, A.C., Jr.; Gubert, P.; Villas Boas, G.R.; Meirelles Paes, M.; Santamaría, A.; Lee, E.; Tinkov, A.A.; Bowman, A.B.; Aschner, M. Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert Rev. Neurother. 2020, 20, 1109–1121. [Google Scholar] [CrossRef]
- Newell, M.E.; Adhikari, S.; Halden, R.U. Systematic and state-of the science review of the role of environmental factors in Amyotrophic Lateral Sclerosis (ALS) or Lou Gehrig’s Disease. Sci. Total Environ. 2022, 817, 152504. [Google Scholar] [CrossRef]
- Bonfiglio, R.; Scimeca, M.; Mauriello, A. The impact of aluminum exposure on human health. Arch. Toxicol. 2023, 97, 2997–2998. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Research 2016, 5, F1000 Faculty Rev-366. [Google Scholar] [CrossRef]
- Zhao, Y.; Ray, A.; Portengen, L.; Vermeulen, R.; Peters, S. Metal Exposure and Risk of Parkinson Disease: A Systematic Review and Meta-Analysis. Am. J. Epidemiol. 2023, 192, 1207–1223. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhou, J.; Zhang, Y.; Huang, H.; Han, J.; Cao, B.; Xu, D.; Zhao, Y.; Chen, G. Risk factors associated with amyotrophic lateral sclerosis based on the observational study: A systematic review and meta-analysis. Front. Neurosci. 2023, 17, 1196722. [Google Scholar] [CrossRef]
- Arab, A.; Mostafalou, S. Neurotoxicity of pesticides in the context of CNS chronic diseases. Int. J. Environ. Health Res. 2022, 32, 2718–2755. [Google Scholar] [CrossRef]
- Vellingiri, B.; Chandrasekhar, M.; Sri Sabari, S.; Gopalakrishnan, A.V.; Narayanasamy, A.; Venkatesan, D.; Iyer, M.; Kesari, K.; Dey, A. Neurotoxicity of pesticides—A link to neurodegeneration. Ecotoxicol. Environ. Saf. 2022, 243, 113972. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Neuropathological Mechanisms Associated with Pesticides in Alzheimer’s Disease. Toxics 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 2017, 61, 101–130. [Google Scholar] [CrossRef]
- Roy, R.; D’Angiulli, A. Air pollution and neurological diseases; current state highlights. Front. Neurosci. 2024, 18, 1351721. [Google Scholar] [CrossRef]
- Fu, C.; Kuang, D.; Zhang, H.; Ren, J.; Chen, J. Different components of air pollutants and neurological disorders. Front. Public Health 2022, 10, 959921. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Cole, T.B.; Dao, K.; Chang, Y.C.; Coburn, J.; Garrick, J.M. Effects of air pollution on the nervous system and its possible role in neurodevelopmental and neurodegenerative disorders. Pharmacol. Ther. 2020, 210, 107523. [Google Scholar] [CrossRef]
- Cheng, S.; Jin, Y.; Dou, Y.; Zhao, Y.; Duan, Y.; Pei, H.; Lyu, P. Long-term particulate matter 2.5 exposure and dementia: A systematic review and meta-analysis. Public Health 2022, 212, 33–41. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, X.; Zhao, X.; Chang, H.; Zhang, J.; Gao, Z.; Mi, Y.; Chen, Y.; Zhang, H.; Huang, C.; et al. Global ambient particulate matter pollution and neurodegenerative disorders: A systematic review of literature and meta-analysis. Environ. Sci. Pollut. Res. Int. 2023, 30, 39418–39430. [Google Scholar] [CrossRef]
- Yu, Z.; Peters, S.; van Boxmeer, L.; Downward, G.S.; Hoek, G.; Kioumourtzoglou, M.A.; Weisskopf, M.G.; Hansen, J.; van den Berg, L.H.; Vermeulen, R.C.H. Long-Term Exposure to Ultrafine Particles and Particulate Matter Constituents and the Risk of Amyotrophic Lateral Sclerosis. Environ. Health Perspect. 2021, 129, 97702. [Google Scholar] [CrossRef]
- Malek, A.M.; Arena, V.C.; Song, R.; Whitsel, E.A.; Rager, J.R.; Stewart, J.; Yanosky, J.D.; Liao, D.; Talbott, E.O. Long-term air pollution and risk of amyotrophic lateral sclerosis mortality in the Women’s Health Initiative cohort. Environ. Res. 2023, 216, 114510. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Stommel, E.W.; Torres-Jardón, R.; Hernández-Luna, J.; Aiello-Mora, M.; González-Maciel, A.; Reynoso-Robles, R.; Pérez-Guillé, B.; Silva-Pereyra, H.G.; Tehuacanero-Cuapa, S.; et al. Alzheimer and Parkinson diseases; frontotemporal lobar degeneration and amyotrophic lateral sclerosis overlapping neuropathology start in the first two decades of life in pollution exposed urbanites and brain ultrafine particulate matter and industrial nanoparticles; including Fe, Ti, Al, V, Ni, Hg, Co, Cu, Zn, Ag, Pt, Ce, La, Pr and W are key players. Metropolitan Mexico City health crisis is in progress. Front. Hum. Neurosci. 2024, 17, 1297467. [Google Scholar]
- Calderón-Garcidueñas, L.; Stommel, E.W.; Lachmann, I.; Waniek, K.; Chao, C.K.; González-Maciel, A.; García-Rojas, E.; Torres-Jardón, R.; Delgado-Chávez, R.; Mukherjee, P.S. TDP-43 CSF Concentrations Increase Exponentially with Age in Metropolitan Mexico City Young Urbanites Highly Exposed to PM2.5 and Ultrafine Particles and Historically Showing Alzheimer and Parkinson’s Hallmarks. Brain TDP-43 Pathology in MMC Residents Is Associated with High Cisternal CSF TDP-43 Concentrations. Toxics 2022, 10, 559. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef]
- He, B.; Zhang, C.; Zhang, X.; Fan, Y.; Zeng, H.; Liu, J.; Meng, H.; Bai, D.; Peng, J.; Zhang, Q.; et al. Tissue-specific 5-hydroxymethylcytosine landscape of the human genome. Nat. Commun. 2021, 12, 4249. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppedè, F. Mitochondrial DNA Methylation and Human Diseases. Int. J. Mol. Sci. 2021, 22, 4594. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Ren, L.; Zhang, C.; Miao, K.; Tan, K.; Yang, Q.; Hu, Y.; Xi, G.; Luo, G.; Yang, M.; et al. Mitochondrial genome undergoes de novo DNA methylation that protects mtDNA against oxidative damage during the peri-implantation window. Proc. Natl. Acad. Sci. USA 2022, 119, e2201168119. [Google Scholar] [CrossRef]
- Sharma, N.; Pasala, M.S.; Prakash, A. Mitochondrial DNA: Epigenetics and environment. Environ. Mol. Mutagen. 2019, 60, 668–682. [Google Scholar] [CrossRef]
- Li, Y. Modern epigenetics methods in biological research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef]
- Lossi, L.; Castagna, C.; Merighi, A. An Overview of the Epigenetic Modifications in the Brain under Normal and Pathological Conditions. Int. J. Mol. Sci. 2024, 25, 3881. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xiao, Y.; Cheng, Y.; Huang, J.; Wei, Q.; Li, C.; Shang, H. Epigenetic clocks in neurodegenerative diseases: A systematic review. J. Neurol. Neurosurg. Psychiatry 2023, 94, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Basha, M.R.; Brock, B.; Cox, D.P.; Cardozo-Pelaez, F.; McPherson, C.A.; Harry, J.; Rice, D.C.; Maloney, B.; Chen, D.; et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): Evidence for a developmental origin and environmental link for AD. J. Neurosci. 2008, 28, 3–9. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppedè, F. Role of epigenetics in Alzheimer’s disease pathogenesis. Neurodegener. Dis. Manag. 2018, 8, 181–193. [Google Scholar] [CrossRef]
- Smith, R.G.; Pishva, E.; Shireby, G.; Smith, A.R.; Roubroeks, J.A.Y.; Hannon, E.; Wheildon, G.; Mastroeni, D.; Gasparoni, G.; Riemenschneider, M.; et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat. Commun. 2021, 12, 3517. [Google Scholar] [CrossRef]
- Villa, C.; Stoccoro, A. Epigenetic Peripheral Biomarkers for Early Diagnosis of Alzheimer’s Disease. Genes 2022, 13, 1308. [Google Scholar] [CrossRef]
- Wüllner, U.; Kaut, O.; deBoni, L.; Piston, D.; Schmitt, I. DNA methylation in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 108–120. [Google Scholar] [CrossRef]
- Song, H.; Chen, J.; Huang, J.; Sun, P.; Liu, Y.; Xu, L.; Wei, C.; Mu, X.; Lu, X.; Wang, W.; et al. Epigenetic modification in Parkinson’s disease. Front. Cell Dev. Biol. 2023, 11, 1123621. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liang, J.; Guo, W.; Zhang, Y.; Wu, X.; Zhang, W. Integrative analysis of DNA methylation and gene expression data for the diagnosis and underlying mechanism of Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 971528. [Google Scholar] [CrossRef]
- Kia, D.A.; Zhang, D.; Guelfi, S.; Manzoni, C.; Hubbard, L.; Reynolds, R.H.; Botía, J.; Ryten, M.; Ferrari, R.; Lewis, P.A.; et al. Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study; Expression; and Epigenetic Data Sets. JAMA Neurol. 2021, 78, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef]
- Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA Methylation Occur in Spinal Cord and Skeletal Muscle of Human SOD1 Mouse Models of ALS and in Human ALS: Targeting DNA Methylation Is Therapeutic. Cells 2022, 11, 3448. [Google Scholar] [CrossRef]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.F.A.; Dekker, A.M.; Westeneng, H.J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic; inflammatory; and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef] [PubMed]
- Tarr, I.S.; McCann, E.P.; Benyamin, B.; Peters, T.J.; Twine, N.A.; Zhang, K.Y.; Zhao, Q.; Zhang, Z.H.; Rowe, D.B.; Nicholson, G.A.; et al. Monozygotic twins and triplets discordant for amyotrophic lateral sclerosis display differential methylation and gene expression. Sci. Rep. 2019, 9, 8254. [Google Scholar] [CrossRef]
- Tremolizzo, L.; Messina, P.; Conti, E.; Sala, G.; Cecchi, M.; Airoldi, L.; Pastorelli, R.; Pupillo, E.; Bandettini Di Poggio, M.; Filosto, M.; et al. Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 98–105. [Google Scholar] [CrossRef]
- Coppedè, F.; Stoccoro, A.; Mosca, L.; Gallo, R.; Tarlarini, C.; Lunetta, C.; Marocchi, A.; Migliore, L.; Penco, S. Increase in DNA methylation in patients with amyotrophic lateral sclerosis carriers of not fully penetrant SOD1 mutations. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 93–101. [Google Scholar] [CrossRef]
- Yang, T.; Li, C.; Wei, Q.; Pang, D.; Cheng, Y.; Huang, J.; Lin, J.; Xiao, Y.; Jiang, Q.; Wang, S.; et al. Genome-wide DNA methylation analysis related to ALS patient progression and survival. J. Neurol. 2024, 271, 2672–2683. [Google Scholar] [CrossRef]
- Reis, A.H.O.; Figalo, L.B.; Orsini, M.; Lemos, B. The implications of DNA methylation for amyotrophic lateral sclerosis. An. Acad. Bras. Cienc. 2023, 95, e20230277. [Google Scholar] [CrossRef] [PubMed]
- Bianchessi, V.; Vinci, M.C.; Nigro, P.; Rizzi, V.; Farina, F.; Capogrossi, M.C.; Pompilio, G.; Gualdi, V.; Lauri, A. Methylation profiling by bisulfite sequencing analysis of the mtDNA Non-Coding Region in replicative and senescent Endothelial Cells. Mitochondrion 2016, 27, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Mawlood, S.K.; Dennany, L.; Watson, N.; Dempster, J.; Pickard, B.S. Quantification of global mitochondrial DNA methylation levels and inverse correlation with age at two CpG sites. Aging 2016, 8, 636–641. [Google Scholar] [CrossRef]
- D’Aquila, P.; Giordano, M.; Montesanto, A.; De Rango, F.; Passarino, G.; Bellizzi, D. Age-and gender-related pattern of methylation in the MT-RNR1 gene. Epigenomics 2015, 7, 707–716. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef]
- Wong, M.; Gertz, B.; Chestnut, B.A.; Martin, L.J. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell. Neurosci. 2013, 7, 279. [Google Scholar] [CrossRef]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppedè, F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced mitochondrial D-loop methylation levels in sporadic amyotrophic lateral sclerosis. Clin. Epigenetics 2020, 12, 137. [Google Scholar] [CrossRef]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Lunnon, K.; Migliore, L.; Coppedè, F. Mitochondrial D-loop methylation levels inversely correlate with disease duration in amyotrophic lateral sclerosis. Epigenomics 2024, 16, 203–214. [Google Scholar] [CrossRef]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered Mitochondrial DNA Methylation Pattern in Alzheimer Disease-Related Pathology and in Parkinson Disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, L.; Han, M.; Liu, X.; Li, F.; Zhou, X.; Wang, Y.; Bi, J. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an APP/PS1 transgenic mouse model of Alzheimer disease. Biochem. Biophys. Res. Commun. 2019, 520, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Siciliano, G.; Migliore, L.; Coppedè, F. Decreased Methylation of the Mitochondrial D-Loop Region in Late-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2017, 59, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Baldacci, F.; Ceravolo, R.; Giampietri, L.; Tognoni, G.; Siciliano, G.; Migliore, L.; Coppedè, F. Increase in Mitochondrial D-Loop Region Methylation Levels in Mild Cognitive Impairment Individuals. Int. J. Mol. Sci. 2022, 23, 5393. [Google Scholar] [CrossRef]
- Lüth, T.; Wasner, K.; Klein, C.; Schaake, S.; Tse, R.; Pereira, S.L.; Laß, J.; Sinkkonen, L.; Grünewald, A.; Trinh, J. Nanopore Single-Molecule Sequencing for Mitochondrial DNA Methylation Analysis: Investigating Parkin-Associated Parkinsonism as a Proof of Concept. Front. Aging Neurosci. 2021, 13, 713084. [Google Scholar] [CrossRef] [PubMed]
- Guitton, R.; Dölle, C.; Alves, G.; Ole-Bjørn, T.; Nido, G.S.; Tzoulis, C. Ultra-deep whole genome bisulfite sequencing reveals a single methylation hotspot in human brain mitochondrial DNA. Epigenetics 2022, 17, 906–921. [Google Scholar] [CrossRef]
- Sharma, A.; Schaefer, S.T.; Sae-Lee, C.; Byun, H.M.; Wüllner, U. Elevated serum mitochondrial DNA in females and lack of altered platelet mitochondrial methylation in patients with Parkinson’s disease. Int. J. Neurosci. 2021, 131, 279–282. [Google Scholar] [CrossRef]
- Stoccoro, A.; Smith, A.R.; Baldacci, F.; Del Gamba, C.; Lo Gerfo, A.; Ceravolo, R.; Lunnon, K.; Migliore, L.; Coppedè, F. Mitochondrial D-Loop Region Methylation and Copy Number in Peripheral Blood DNA of Parkinson’s Disease Patients. Genes 2021, 12, 720. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Huang, H.; Wu, J.; Zawia, N.H. Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: Implications for Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Tarale, P.; Sivanesan, S.; Daiwile, A.P.; Stöger, R.; Bafana, A.; Naoghare, P.K.; Parmar, D.; Chakrabarti, T.; Kannan, K. Global DNA methylation profiling of manganese-exposed human neuroblastoma SH-SY5Y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch. Toxicol. 2017, 91, 2629–2641. [Google Scholar] [CrossRef]
- Yang, N.; Wei, Y.; Wang, T.; Guo, J.; Sun, Q.; Hu, Y.; Yan, X.; Zhu, X.; Tang, B.; Xu, Q. Genome-wide analysis of DNA methylation during antagonism of DMOG to MnCl2-induced cytotoxicity in the mouse substantia nigra. Sci. Rep. 2016, 6, 28933. [Google Scholar] [CrossRef]
- Mythri, R.B.; Raghunath, N.R.; Narwade, S.C.; Pandareesh, M.D.R.; Sabitha, K.R.; Aiyaz, M.; Chand, B.; Sule, M.; Ghosh, K.; Kumar, S.; et al. Manganese- and 1-methyl-4-phenylpyridinium-induced neurotoxicity display differences in morphological; electrophysiological and genome-wide alterations: Implications for idiopathic Parkinson’s disease. J. Neurochem. 2017, 143, 334–358. [Google Scholar] [CrossRef] [PubMed]
- da Silva, V.K.; de Freitas, B.S.; Dornelles, V.C.; Kist, L.W.; Bogo, M.R.; Silva, M.C.; Streck, E.L.; Hallak, J.E.; Zuardi, A.W.; Crippa, J.A.S.; et al. Novel insights into mitochondrial molecular targets of iron-induced neurodegeneration: Reversal by cannabidiol. Brain Res. Bull. 2018, 139, 1–8. [Google Scholar] [CrossRef]
- Kanthasamy, A.G.; Kitazawa, M.; Kanthasamy, A.; Anantharam, V. Dieldrin-induced neurotoxicity: Relevance to Parkinson’s disease pathogenesis. Neurotoxicology 2005, 26, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Kochmanski, J.; VanOeveren, S.E.; Patterson, J.R.; Bernstein, A.I. Developmental Dieldrin Exposure Alters DNA Methylation at Genes Related to Dopaminergic Neuron Development and Parkinson’s Disease in Mouse Midbrain. Toxicol. Sci. 2019, 169, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sánchez, E.; Yescas, P.; Rodríguez-Violante, M.; Martínez-Rodríguez, N.; Díaz-López, J.N.; Ochoa, A.; Valdes-Rojas, S.S.; Magos-Rodríguez, D.; Rojas-Castañeda, J.C.; Cervantes-Arriaga, A.; et al. Association of polymorphisms and reduced expression levels of the NR4A2 gene with Parkinson’s disease in a Mexican population. J. Neurol. Sci. 2017, 379, 58–63. [Google Scholar] [CrossRef]
- Laguna, A.; Schintu, N.; Nobre, A.; Alvarsson, A.; Volakakis, N.; Jacobsen, J.K.; Gómez-Galán, M.; Sopova, E.; Joodmardi, E.; Yoshitake, T.; et al. Dopaminergic control of autophagic-lysosomal function implicates Lmx1b in Parkinson’s disease. Nat. Neurosci. 2015, 18, 826–835. [Google Scholar] [CrossRef]
- Kochmanski, J.; Virani, M.; Kuhn, N.C.; Boyd, S.L.; Becker, K.; Adams, M.; Bernstein, A.I. Developmental origins of parkinson’s disease risk: Perinatal exposure to the organochlorine pesticide dieldrin leads to sex-specific DNA modifications in critical neurodevelopmental pathways in the mouse midbrain. Toxicol. Sci. 2024, 201, 263–281. [Google Scholar] [CrossRef]
- Freeman, D.M.; Lou, D.; Li, Y.; Martos, S.N.; Wang, Z. The conserved DNMT1-dependent methylation regions in human cells are vulnerable to neurotoxicant rotenone exposure. Epigenetics Chromatin 2020, 13, 17. [Google Scholar] [CrossRef]
- Qi, Z.X.; Yan, Q.; Fan, X.J.; Peng, J.Y.; Zhu, H.X.; Jiang, Y.M.; Chen, L.; Zhuang, Q.X. Role of HCN channels in the functions of basal ganglia and Parkinson’s disease. Cell. Mol. Life Sci. 2024, 81, 135. [Google Scholar] [CrossRef]
- Krüger, R.; Fischer, C.; Schulte, T.; Strauss, K.M.; Müller, T.; Woitalla, D.; Berg, D.; Hungs, M.; Gobbele, R.; Berger, K.; et al. Mutation analysis of the neurofilament M gene in Parkinson’s disease. Neurosci. Lett. 2003, 351, 125–129. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, L.; Li, H.; Shen, J.; Li, H.; Xing, Y. Novel diagnostic biomarkers related to immune infiltration in Parkinson’s disease by bioinformatics analysis. Front. Neurosci. 2023, 17, 1083928. [Google Scholar] [CrossRef]
- Freeman, D.M.; Wang, Z. Epigenetic Vulnerability of Insulator CTCF Motifs at Parkinson’s Disease-Associated Genes in Response to Neurotoxicant Rotenone. Front. Genet. 2020, 11, 627. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Qu, Q.; Chen, X.; Zhao, L.; Jiao, Z.; Wan, Z.; Kwok, H.F.; Qu, S. Downregulation of Ambra1 by altered DNA methylation exacerbates dopaminergic neuron damage in a fenpropathrin-induced Parkinson-like mouse model. Ecotoxicol. Environ. Saf. 2024, 271, 115995. [Google Scholar] [CrossRef] [PubMed]
- Van Humbeeck, C.; Cornelissen, T.; Vandenberghe, W. Ambra1: A Parkin-binding protein involved in mitophagy. Autophagy 2011, 7, 1555–1556. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, J.; Zhou, Q.; Wang, J.; Liu, C.; Tian, Y.; Huang, D.; Ye, H.; Jin, Y. Exposure to a real traffic environment impairs brain cognition in aged mice. Environ. Res. 2022, 215, 114181. [Google Scholar] [CrossRef] [PubMed]
- Colicino, E.; Just, A.; Kioumourtzoglou, M.A.; Vokonas, P.; Cardenas, A.; Sparrow, D.; Weisskopf, M.; Nie, L.H.; Hu, H.; Schwartz, J.D.; et al. Blood DNA methylation biomarkers of cumulative lead exposure in adults. J. Expo. Sci. Environ. Epidemiol. 2021, 31, 108–116. [Google Scholar] [CrossRef]
- Paul, K.C.; Horvath, S.; Del Rosario, I.; Bronstein, J.M.; Ritz, B. DNA methylation biomarker for cumulative lead exposure is associated with Parkinson’s disease. Clin. Epigenetics 2021, 13, 59. [Google Scholar] [CrossRef]
- Yang, X.; Yuan, Y.; Lu, X.; Yang, J.; Wang, L.; Song, J.; Nie, J.; Zhang, Q.; Niu, Q. The Relationship Between Cognitive Impairment and Global DNA Methylation Decrease Among Aluminum Potroom Workers. J. Occup. Environ. Med. 2015, 57, 713–717. [Google Scholar] [CrossRef]
- Searles Nielsen, S.; Checkoway, H.; Criswell, S.R.; Farin, F.M.; Stapleton, P.L.; Sheppard, L.; Racette, B.A. Inducible nitric oxide synthase gene methylation and parkinsonism in manganese-exposed welders. Park. Relat. Disord. 2015, 21, 355–360. [Google Scholar] [CrossRef]
- Castillo, S.; Muñoz, P.; Behrens, M.I.; Diaz-Grez, F.; Segura-Aguilar, J. On the Role of Mining Exposure in Epigenetic Effects in Parkinson’s Disease. Neurotox. Res. 2017, 32, 172–174. [Google Scholar] [CrossRef]
- Freydenzon, A.; Nabais, M.F.; Lin, T.; Williams, K.L.; Wallace, L.; Henders, A.K.; Blair, I.P.; Wray, N.R.; Pamphlett, R.; McRae, A.F. Association between DNA methylation variability and self-reported exposure to heavy metals. Sci. Rep. 2022, 12, 10582. [Google Scholar] [CrossRef] [PubMed]
- Brusati, A.; Peverelli, S.; Calzari, L.; Tiloca, C.; Casiraghi, V.; Source, M.N.; Invernizzi, S.; Carbone, E.; Cavagnola, R.; Verde, F.; et al. Exploring epigenetic drift and rare epivariations in amyotrophic lateral sclerosis by epigenome-wide association study. Front. Aging Neurosci. 2023, 15, 1272135. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 2009, 4, e6617. [Google Scholar] [CrossRef] [PubMed]
- Furlong, M.A.; Paul, K.C.; Yan, Q.; Chuang, Y.H.; Cockburn, M.G.; Bronstein, J.M.; Horvath, S.; Ritz, B. An epigenome-wide association study of ambient pyrethroid pesticide exposures in California’s central valley. Int. J. Hyg. Environ. Health 2020, 229, 113569. [Google Scholar] [CrossRef]
- Paul, K.C.; Chuang, Y.H.; Cockburn, M.; Bronstein, J.M.; Horvath, S.; Ritz, B. Organophosphate pesticide exposure and differential genome-wide DNA methylation. Sci. Total Environ. 2018, 645, 1135–1143. [Google Scholar] [CrossRef]
- Go, R.C.P.; Corley, M.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.H.; Maunakea, A.K.; He, Q.; Tiirikainen, M.I. Genome-wide epigenetic analyses in Japanese immigrant plantation workers with Parkinson’s disease and exposure to organochlorines reveal possible involvement of glial genes and pathways involved in neurotoxicity. BMC Neurosci. 2020, 21, 31. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Casazza, W.; Artaud, F.; Konwar, C.; Merrill, S.M.; Domenighetti, C.; Schulze-Hentrich, J.M.; Lesage, S.; Brice, A.; Corvol, J.C.; et al. Genetic variation and pesticide exposure influence blood DNA methylation signatures in females with early-stage Parkinson’s disease. NPJ Park. Dis. 2024, 10, 98. [Google Scholar] [CrossRef]
- Li, Z.; Liang, D.; Ebelt, S.; Gearing, M.; Kobor, M.S.; Konwar, C.; Maclsaac, J.L.; Dever, K.; Wingo, A.P.; Levey, A.I.; et al. Differential DNA methylation in the brain as potential mediator of the association between traffic-related PM2.5 and neuropathology markers of Alzheimer’s disease. Alzheimers Dement. 2024, 20, 2538–2551. [Google Scholar] [CrossRef]
- Wang, C.; Amini, H.; Xu, Z.; Peralta, A.A.; Yazdi, M.D.; Qiu, X.; Wei, Y.; Just, A.; Heiss, J.; Hou, L.; et al. Long-term exposure to ambient fine particulate components and leukocyte epigenome-wide DNA Methylation in older men: The Normative Aging Study. Environ. Health 2023, 22, 54. [Google Scholar] [CrossRef]
- Madugundu, G.S.; Cadet, J.; Wagner, J.R. Hydroxyl-radical-induced oxidation of 5-methylcytosine in isolated and cellular DNA. Nucleic Acids Res. 2014, 42, 7450–7460. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics; oxidative stress; and Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Maloney, B. The “LEARn” (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer’s disease; and proposes remedial steps. Exp. Gerontol. 2010, 45, 291–296. [Google Scholar] [CrossRef]
- Dimauro, I.; Paronetto, M.P.; Caporossi, D. Exercise; redox homeostasis and the epigenetic landscape. Redox Biol. 2020, 35, 101477. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Yurov, Y.B.; Vorsanova, S.G.; Kutsev, S.I. Chromosome Instability; Aging and Brain Diseases. Cells 2021, 10, 1256. [Google Scholar] [CrossRef] [PubMed]
- Bustaffa, E.; Stoccoro, A.; Bianchi, F.; Migliore, L. Genotoxic and epigenetic mechanisms in arsenic carcinogenicity. Arch. Toxicol. 2014, 88, 1043–1067. [Google Scholar] [CrossRef]
- Barnett, J.B.; Brundage, K.M. Immunotoxicology of pesticides and chemotherapies. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 467–487. [Google Scholar]
- Reddam, A.; McLarnan, S.; Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: A Review of Recent Literature. Curr. Environ. Health Rep. 2022, 9, 631–649. [Google Scholar] [CrossRef]
- Santos, J.H. Mitochondria signaling to the epigenome: A novel role for an old organelle. Free Radic. Biol. Med. 2021, 170, 59–69. [Google Scholar] [CrossRef]
- Byun, H.M.; Panni, T.; Motta, V.; Hou, L.; Nordio, F.; Apostoli, P.; Bertazzi, P.A.; Baccarelli, A.A. Effects of airborne pollutants on mitochondrial DNA methylation. Part. Fibre Toxicol. 2013, 10, 18. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Hedmer, M.; Hossain, M.B.; Tinnerberg, H.; Broberg, K.; Albin, M. Occupational exposure to particles and mitochondrial DNA—Relevance for blood pressure. Environ. Health 2017, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.G.; Byun, H.M.; Gyselaers, W.; Lefebvre, W.; Baccarelli, A.A.; Nawrot, T.S. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics 2015, 10, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.M.; Colicino, E.; Trevisi, L.; Fan, T.; Christiani, D.C.; Baccarelli, A.A. Effects of Air Pollution and Blood Mitochondrial DNA Methylation on Markers of Heart Rate Variability. J. Am. Heart Assoc. 2016, 5, e003218. [Google Scholar] [CrossRef] [PubMed]
- Seow, W.J.; Hu, W.; Dai, Y.; Vermeulen, R.; Byun, H.M.; Wong, J.Y.Y.; Bassig, B.A.; Blechter, B.; Duan, H.; Niu, Y.; et al. Association between diesel exhaust exposure and mitochondrial DNA methylation. Carcinogenesis 2022, 43, 1131–1136. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, J.; Guo, L.; Lan, Y.; Li, G.; Liu, Q.; Li, H.; Deng, F.; Guo, X.; Wu, S. Short-term effects of ambient gaseous air pollution on blood platelet mitochondrial DNA methylation and myocardial ischemia. Environ. Int. 2024, 185, 108533. [Google Scholar] [CrossRef]
- Scott, L.L.; Downing, T.G. A Single Neonatal Exposure to BMAA in a Rat Model Produces Neuropathology Consistent with Neurodegenerative Diseases. Toxins 2017, 10, 22. [Google Scholar] [CrossRef]
- Longinetti, E.; Pupillo, E.; Belometti, C.; Bianchi, E.; Poloni, M.; Fang, F.; Beghi, E. Geographical clusters of amyotrophic lateral sclerosis and the Bradford Hill criteria. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 329–343. [Google Scholar] [CrossRef]
- Pierozan, P.; Cattani, D.; Karlsson, O. Hippocampal neural stem cells are more susceptible to the neurotoxin BMAA than primary neurons: Effects on apoptosis, cellular differentiation, neurite outgrowth, and DNA methylation. Cell Death Dis. 2020, 11, 910. [Google Scholar] [CrossRef]
- Bucher, M.L.; Anderson, F.L.; Lai, Y.; Dicent, J.; Miller, G.W.; Zota, A.R. Exposomics as a tool to investigate differences in health and disease by sex and gender. Exposome 2023, 3, osad003. [Google Scholar] [CrossRef]
- Gauvrit, T.; Benderradji, H.; Buée, L.; Blum, D.; Vieau, D. Early-Life Environment Influence on Late-Onset Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 834661. [Google Scholar] [CrossRef]

{kind=link}
| Experimental Model | Disease Model | Epigenetic Target | Results | Reference |
|---|---|---|---|---|
| Metals | ||||
| Cortical brain tissue of monkeys exposed to lead as infants, which later developed AD-like pathology by the age of 23 | AD | DNMT1 activity | Infantile exposure to lead induced increased expression of APP and BACE1 with concomitant decreased activity of DNMT1 in aged monkeys (23 years old) | [67] |
| Cortical brain tissue of monkeys exposed to lead as infants, which later developed AD-like pathology by the age of 23 | AD | Dnmts, Mecp2, histone-modifying proteins | Infantile exposure to lead induced increased expression of neurobiology-related genes and decreased expression of Dnmts, MeCP2, and proteins involved in histone modifications | [99] |
| Human neuroblastoma (SH-SY5Y) cells exposed to manganese as a model of idiopathic PD | PD | Genome-wide DNA methylation | DNA methylation alterations in genes involved in the onset of PD, including hypermethylation of PINK1, PARK2, and TH | [100] |
| Substantia nigra samples of mice exposed to manganese as a model of PD | PD | Genome-wide DNA methylation | DNA methylation in the promoter region of 226 genes involved in mitochondrial function, cell cycle, DNA damage response, and ion transportation was found to be regulated (mainly hypermethylated) by manganese treatment | [101] |
| Rat dopaminergic cells treated with manganese and MPP+ as models of PD | PD | Genome-wide DNA methylation | Manganese mainly induced hypermethylation in genes involved in cell differentiation and lipid metabolism. MPP+ mainly induced hypermethylation in genes involved in mitochondrial function, autophagy/mitophagy, and WNT signaling | [102] |
| Hippocampal specimens from rats orally exposed to iron levels that cause memory impairment | Neurodegeneration | Global 5-mC and 5-hmC mtDNA content | Iron exposure during the neonatal period at doses that induced neurodegenerative processes led to decreased 5-mC and 5-hmC levels in mtDNA in adulthood | [103] |
| Pesticides | ||||
| Substantia nigra of mice developmentally exposed to dieldrin to model an early stage of PD | PD | Genome-wide DNA methylation | Developmental dieldrin exposure induced altered methylation levels in several genes, including hypermethylation of Nr4a2 and hypomethylation of Lmx1b which are involved in dopaminergic neuron development and maintenance | [105] |
| Substantia nigra of mice developmentally exposed to dieldrin to model an early stage of PD | PD | Methyl-sequencing of targeted regions | Dieldrin induced DNA methylation changes in pup mice exposed to the pesticide in utero and after birth until 36 weeks of age in a sex-specific manner in loci associated with pathways related to neurodevelopment, dopaminergic neuron differentiation, synaptogenesis, and synaptic plasticity | [108] |
| Human embryonic kidney cell with a neuronal lineage phenotype treated with rotenone as a model of PD | PD | Genome-wide DNA methylation | Rotenone treatment induced hypomethylation of genes involved in neuronal development and maturation, including HCN2 and NEFM | [109] |
| Human embryonic kidney cell with a neuronal lineage phenotype treated with rotenone as a model of PD | PD | Genome-wide methylation | Altered methylation in 45 CpG sites (53% hypermethylated) surrounding CTCF binding sites in 7 PD-associated genes, including BMP4, UBOX5, GPRIN3, FER, CNKSR3, PARK2, and CHCHD2 | [113] |
| Midbrain of mice exposed to fenpropathrin as a model of PD | PD | Genome-wide DNA methylation | Hypermethylation of the Ambra1 gene, which in turn was downregulated, led to dopaminergic neuron damage through the Ambra1/Parkin/LC3B-mediated mitophagy pathway | [114] |
| Air pollution | ||||
| Hippocampus samples of aged mice exposed to traffic-related air pollution which developed impairment in memory function | AD | Abca7 and Pyk2 genes | Increased methylation levels of Abca7 and decreased methylation of Pyk2 genes, together with altered mRNA expression levels | [116] |
| Model | Exposure | Disease | Epigenetic Target | Results | Reference |
|---|---|---|---|---|---|
| Metals | |||||
| Peripheral blood of PD (n = 1528) and control subjects (n = 1169) | Lead | PD | Genome-wide DNA methylation | PD patients had increased DNA methylation-predicted lead levels in tibial bone | [118] |
| Peripheral blood of 366 Al potroom workers, including 43 MCI | Aluminium | MCI | Global DNA methylation | Increased aluminum serum levels were inversely correlated with global DNA methylation and MMSE | [119] |
| Peripheral blood of 201 workers, including 49 with parkinsonism | Manganese | PD | NOS2 gene methylation | Workers with parkinsonism had lower mean methylation levels of NOS2, a gene involved in inflammation | [120] |
| Peripheral blood samples of 45 PD patients and 52 control subjects | Heavy metal mining | PD | Global 5-mC | Decrease in global DNA methylation in PD patients either exposed or not exposed to mining activity. No difference between exposed and non-exposed PD patients | [121] |
| Peripheral blood samples of 438 ALS cases and 417 controls | Cadmium, mercury, and metallurgy | ALS | Genome-wide DNA methylation | Self-reported cadmium, mercury, and metallurgy exposure was associated with methylation levels of seven CpG sites across the genome in ALS, including hypermethylation of GNRHR2, PEX11B, ZFR2, LINGO1, and PRKG1-AS1 and hypomethylation of P2RY6 and KSR2 genes | [122] |
| Peripheral blood of 61 ALS patients and 61 controls | Various chemicals, metals, pesticides, and air pollutants | ALS | Genome-wide DNA methylation | ALS epigenetic signature associated with exposure to various metals, including sodium arsenite and nickel, air pollutants, including PM, and pesticides, including rotenone | [123] |
| Pesticides | |||||
| Brain tissue from a pair of monozygotic twins discordant for AD | Pesticides | AD | Global DNA methylation | Global DNA hypomethylation in the AD twin who worked in contact with pesticides | [124] |
| Peripheral blood DNA of 237 individuals | Pesticide (pyrethroid) | Control subjects | Genome-wide DNA methylation | Several CpG sites were associated with pyrethroid exposure, some in genes related to AD, PD, and ALS pathology, including the RTN3 and TMOD3 genes | [125] |
| Peripheral blood samples of 342 PD patients and 238 controls | Pesticides (organophosphate insecticides) | PD | Genome-wide DNA methylation | 70 CpG sites were associated with pesticide exposure, of which 7 were specific to PD patients, such as the MYH15, MFAP2, and KIAA0319 genes | [126] |
| Matched peripheral blood and post-mortem brain in 20 PD cases | Pesticides | PD | Genome-wide DNA methylation | By comparing individuals exposed more than 10 years and 0 years, 7 and 123 DML in brain and blood DNA, respectively, were identified. DML were mainly associated with genes involved in neurotoxic and neuropathologic pathways | [127] |
| Peripheral blood of agricultural workers, including 71 early-stage PD cases and 147 control subjects | Pesticides | PD | Genome-wide DNA methylation | Pesticide exposure influenced blood DNA methylation in females at the early stages of PD, in various genes including the NFATC1 and DLGAP1 genes | [128] |
| Air pollution | |||||
| Prefrontal cortex tissue of 159 donors evaluated for AD-related neuropathological markers | PM2.5 | AD | Genome-wide DNA methylation | PM2.5 exposure induced altered DNA methylation of twenty-four CpG sites that were associated with neuropathology markers of AD. Several CpG sites were located in genes related to neuroinflammation, including SORBS2, PDE11A, and GABBR1 | [129] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoccoro, A.; Coppedè, F. Exposure to Metals, Pesticides, and Air Pollutants: Focus on Resulting DNA Methylation Changes in Neurodegenerative Diseases. Biomolecules 2024, 14, 1366. https://doi.org/10.3390/biom14111366
Stoccoro A, Coppedè F. Exposure to Metals, Pesticides, and Air Pollutants: Focus on Resulting DNA Methylation Changes in Neurodegenerative Diseases. Biomolecules. 2024; 14(11):1366. https://doi.org/10.3390/biom14111366
Chicago/Turabian StyleStoccoro, Andrea, and Fabio Coppedè. 2024. "Exposure to Metals, Pesticides, and Air Pollutants: Focus on Resulting DNA Methylation Changes in Neurodegenerative Diseases" Biomolecules 14, no. 11: 1366. https://doi.org/10.3390/biom14111366
APA StyleStoccoro, A., & Coppedè, F. (2024). Exposure to Metals, Pesticides, and Air Pollutants: Focus on Resulting DNA Methylation Changes in Neurodegenerative Diseases. Biomolecules, 14(11), 1366. https://doi.org/10.3390/biom14111366