
Macrophages in Calcific Aortic Valve Disease: Paracrine and Juxtacrine Disease Drivers
 , , and
, , and 
{kind=link}
{kind=link}
Abstract
1. Introduction
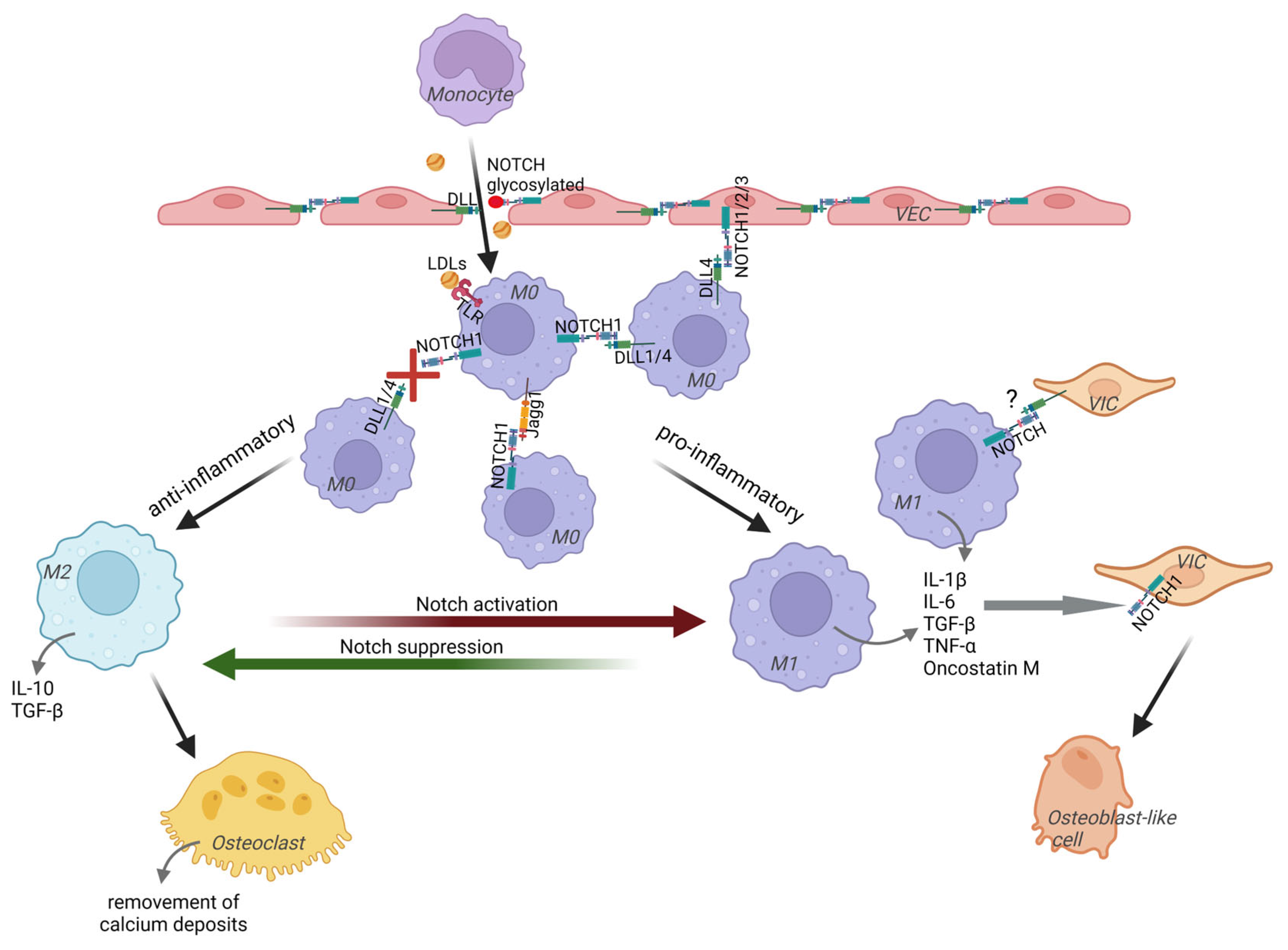
2. Notch in Driving Calcific Aortic Valve Disease
3. Macrophages Play a Role in CAVD Progression in Different Ways, Depending on Their Type
4. Macrophages Play a Signaling Role in Resident Valvular Cells Transformations or Directly Affect the Calcification Process
5. Macrophages in CAVD Have Strong Pro-Inflammatory Properties
5.1. Notch Signaling in Macrophages Regulates Their Pro-Inflammatory Activity
5.2. Accumulated Lipoproteins Influence on Macrophages’ Inflammatory Potential
5.3. The Increase in Inflammatory Activity Occurs Even at the Monocyte Stage
5.4. Increase in Pro-Inflammatory Properties at the Stage of Hematopoietic Precursors
5.4.1. Clonal Hematopoiesis of Indeterminate Potential
5.4.2. Trained Immunity and CAVD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Butcher, J.T.; Mahler, G.J.; Hockaday, L.A. Aortic Valve Disease and Treatment: The Need for Naturally Engineered Solutions. Adv. Drug Deliv. Rev. 2011, 63, 242–268. [Google Scholar] [CrossRef] [PubMed]
- Malashicheva, A.; Kostina, A.; Kostareva, A.; Irtyuga, O.; Gordeev, M.; Uspensky, V. Notch Signaling in the Pathogenesis of Thoracic Aortic Aneurysms: A Bridge between Embryonic and Adult States. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165631. [Google Scholar] [CrossRef] [PubMed]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH Regulation of the Endothelial Cell Phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef]
- Derada Troletti, C.; Lopes Pinheiro, M.A.; Charabati, M.; Gowing, E.; van het Hof, B.; van der Pol, S.M.A.; Geerts, D.; Prat, A.; Fontijn, R.D.; Unger, W.W.; et al. Notch Signaling Is Impaired during Inflammation in a Lunatic Fringe-Dependent Manner. Brain Behav. Immun. 2018, 69, 48–56. [Google Scholar] [CrossRef]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/β-Catenin Pathway Modulates Vascular Remodeling and Specification by Upregulating Dll4/Notch Signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef]
- Zeng, Q.; Song, R.; Ao, L.; Xu, D.; Venardos, N.; Fullerton, D.A.; Meng, X. Augmented Osteogenic Responses in Human Aortic Valve Cells Exposed to OxLDL and TLR4 Agonist: A Mechanistic Role of Notch1 and NF-ΚB Interaction. PLoS ONE 2014, 9, e95400. [Google Scholar] [CrossRef]
- O’Brien, K.D.; Reichenbach, D.D.; Marcovina, S.M.; Kuusisto, J.; Alpers, C.E.; Otto, C.M. Apolipoproteins B, (a), and E Accumulate in the Morphologically Early Lesion of ‘Degenerative’ Valvular Aortic Stenosis. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 523–532. [Google Scholar] [CrossRef]
- Sucosky, P.; Balachandran, K.; Elhammali, A.; Jo, H.; Yoganathan, A.P. Altered Shear Stress Stimulates Upregulation of Endothelial VCAM-1 and ICAM-1 in a BMP-4– and TGF-Β1–Dependent Pathway. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 254–260. [Google Scholar] [CrossRef]
- Ghaisas, N.K.; Foley, J.B.; O’Briain, D.S.; Crean, P.; Kelleher, D.; Walsh, M. Adhesion Molecules in Nonrheumatic Aortic Valve Disease: Endothelial Expression, Serum Levels and Effects of Valve Replacement. J. Am. Coll. Cardiol. 2000, 36, 2257–2262. [Google Scholar] [CrossRef]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J. Effect of Lipid Lowering with Rosuvastatin on Progression of Aortic Stenosis. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Kuusisto, J.; Reichenbach, D.D.; Gown, A.M.; O’Brien, K.D. Characterization of the Early Lesion of “degenerative” Valvular Aortic Stenosis. Histological and Immunohistochemical Studies. Circulation 1994, 90, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Wei, S.; Schwertani, A. A Notch More: Molecular Players in Bicuspid Aortic Valve Disease. J. Mol. Cell Cardiol. 2019, 134, 62–68. [Google Scholar] [CrossRef]
- Tie, R.; Li, H.; Cai, S.; Liang, Z.; Shan, W.; Wang, B.; Tan, Y.; Zheng, W.; Huang, H. Interleukin-6 Signaling Regulates Hematopoietic Stem Cell Emergence. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Song, Y.; Su, R.-W.; Joshi, N.R.; Kim, T.H.; Lessey, B.A.; Jeong, J.-W.; Fazleabas, A.T. Interleukin-6 (IL-6) Activates the NOTCH1 Signaling Pathway Through E-Proteins in Endometriotic Lesions. J. Clin. Endocrinol. Metab. 2020, 105, 1316–1326. [Google Scholar] [CrossRef]
- Kostina, A.S.; Uspensky, V.E.; Irtyuga, O.B.; Ignatieva, E.V.; Freylikhman, O.; Gavriliuk, N.D.; Moiseeva, O.M.; Zhuk, S.; Tomilin, A.; Kostareva, A.A.; et al. Notch-Dependent EMT Is Attenuated in Patients with Aortic Aneurysm and Bicuspid Aortic Valve. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 733–740. [Google Scholar] [CrossRef]
- Kostina, A.; Shishkova, A.; Ignatieva, E.; Irtyuga, O.; Bogdanova, M.; Levchuk, K.; Golovkin, A.; Zhiduleva, E.; Uspenskiy, V.; Moiseeva, O.; et al. Different Notch Signaling in Cells from Calcified Bicuspid and Tricuspid Aortic Valves. J. Mol. Cell Cardiol. 2018, 114, 211–219. [Google Scholar] [CrossRef]
- Yip, C.Y.Y.; Blaser, M.C.; Mirzaei, Z.; Zhong, X.; Simmons, C.A. Inhibition of Pathological Differentiation of Valvular Interstitial Cells by C-Type Natriuretic Peptide. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1881–1889. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 Cause Aortic Valve Disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Deregowski, V.; Gazzerro, E.; Priest, L.; Rydziel, S.; Canalis, E. Notch 1 Overexpression Inhibits Osteoblastogenesis by Suppressing Wnt/β-Catenin but Not Bone Morphogenetic Protein Signaling. J. Biol. Chem. 2006, 281, 6203–6210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Sweetwyne, M.T.; Hankenson, K.D. PKCδ Is Required for Jagged-1 Induction of Human Mesenchymal Stem Cell Osteogenic Differentiation. Stem Cells 2013, 31, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ryzhova, L.M.; Sewell-Loftin, M.K.; Brown, C.B.; Huppert, S.S.; Baldwin, H.S.; Merryman, W.D. Notch1 Mutation Leads to Valvular Calcification Through Enhanced Myofibroblast Mechanotransduction. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Hans, C.P.; Koenig, S.N.; Nichols, H.A.; Galindo, C.L.; Garner, H.R.; Merrill, W.H.; Hinton, R.B.; Garg, V. Inhibitory Role of Notch1 in Calcific Aortic Valve Disease. PLoS ONE 2011, 6, e27743. [Google Scholar] [CrossRef]
- Li, G.; Shen, N.; Deng, H.; Wang, Y.; Kong, G.; Shi, J.; Dong, N.; Deng, C. Abnormal Mechanical Stress on Bicuspid Aortic Valve Induces Valvular Calcification and Inhibits Notch1/NICD/Runx2 Signal. PeerJ 2023, 11, e14950. [Google Scholar] [CrossRef]
- Majumdar, U.; Manivannan, S.; Basu, M.; Ueyama, Y.; Blaser, M.C.; Cameron, E.; McDermott, M.R.; Lincoln, J.; Cole, S.E.; Wood, S.; et al. Nitric Oxide Prevents Aortic Valve Calcification by S-Nitrosylation of USP9X to Activate NOTCH Signaling. Sci. Adv. 2021, 7, eabe3706. [Google Scholar] [CrossRef]
- Semenova, D.; Bogdanova, M.; Kostina, A.; Golovkin, A.; Kostareva, A.; Malashicheva, A. Dose-Dependent Mechanism of Notch Action in Promoting Osteogenic Differentiation of Mesenchymal Stem Cells. Cell Tissue Res. 2020, 379, 169–179. [Google Scholar] [CrossRef]
- New, S.E.P.; Aikawa, E. Cardiovascular Calcification-An Inflammatory Disease. Circ. J. 2011, 75, 1305–1313. [Google Scholar] [CrossRef]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.-L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis Associates with Inflammation in Early-Stage Atherosclerosis Evaluated by Molecular Imaging In Vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef]
- Hinz, B. Formation and Function of the Myofibroblast during Tissue Repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Yang, X.; Meng, X.; Su, X.; Mauchley, D.C.; Ao, L.; Cleveland, J.C.; Fullerton, D.A. Bone Morphogenic Protein 2 Induces Runx2 and Osteopontin Expression in Human Aortic Valve Interstitial Cells: Role of Smad1 and Extracellular Signal-Regulated Kinase 1/2. J. Thorac. Cardiovasc. Surg. 2009, 138, 1008–1015.e1. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, A.; Bravou, V.; Peroukides, S.; Kaklamanis, L.; Varakis, J.; Alexopoulos, D.; Papadaki, H. Bone Regulatory Factors NFATc1 and Osterix in Human Calcific Aortic Valves. Int. J. Cardiol. 2010, 139, 142–149. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, D.; Boulanger, M.-C.; Mahmut, A.; Bouchareb, R.; Laflamme, M.-H.; Fournier, D.; Pibarot, P.; Bossé, Y.; Mathieu, P. P2Y2 Receptor Represses IL-6 Expression by Valve Interstitial Cells through Akt: Implication for Calcific Aortic Valve Disease. J. Mol. Cell Cardiol. 2014, 72, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Torre, M.; Hwang, D.H.; Padera, R.F.; Mitchell, R.N.; VanderLaan, P.A. Osseous and Chondromatous Metaplasia in Calcific Aortic Valve Stenosis. Cardiovasc. Pathol. 2016, 25, 18–24. [Google Scholar] [CrossRef]
- Mohler, E.R.; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone Formation and Inflammation in Cardiac Valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef]
- Goody, P.R.; Hosen, M.R.; Christmann, D.; Niepmann, S.T.; Zietzer, A.; Adam, M.; Bönner, F.; Zimmer, S.; Nickenig, G.; Jansen, F. Aortic Valve Stenosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 885–900. [Google Scholar] [CrossRef]
- Lee, Y.S.; Chou, Y.Y. Pathogenetic Mechanism of Senile Calcific Aortic Stenosis: The Role of Apoptosis. Chin. Med. J. 1998, 111, 934–939. [Google Scholar]
- Jian, B.; Narula, N.; Li, Q.; Mohler, E.R.; Levy, R.J. Progression of Aortic Valve Stenosis: TGF-Β1 Is Present in Calcified Aortic Valve Cusps and Promotes Aortic Valve Interstitial Cell Calcification via Apoptosis. Ann. Thorac. Surg. 2003, 75, 457–465. [Google Scholar] [CrossRef]
- Bonetti, A.; Marchini, M.; Ortolani, F. Ectopic Mineralization in Heart Valves: New Insights from in Vivo and in Vitro Procalcific Models and Promising Perspectives on Noncalcifiable Bioengineered Valves. J. Thorac. Dis. 2019, 11, 2126–2143. [Google Scholar] [CrossRef]
- Kim, K.M. Apoptosis and Calcification. Scanning Microsc. 1995, 9, 1137–1175; discussion 1175–1178. [Google Scholar]
- Kim, K.M. Calcification of Matrix Vesicles in Human Aortic Valve and Aortic Media. Fed. Proc. 1976, 35, 156–162. [Google Scholar] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification In Vitro. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- New, S.E.P.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-Derived Matrix Vesicles. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-F.; Wang, Y.; Zheng, D.-D.; Xu, H.-X.; Wang, T.; Pan, M.; Shi, J.-H.; Zhu, J.-H. M1 Macrophages Promote Aortic Valve Calcification Mediated by MicroRNA-214/TWIST1 Pathway in Valvular Interstitial Cells. Am. J. Transl. Res. 2016, 8, 5773–5783. [Google Scholar]
- Klauzen, P.; Semenova, D.; Kostina, D.; Uspenskiy, V.; Malashicheva, A. Purinergic Signaling in Pathologic Osteogenic Differentiation of Aortic Valve Interstitial Cells from Patients with Aortic Valve Calcification. Biomedicines 2023, 11, 307. [Google Scholar] [CrossRef]
- Hulin, A.; Anstine, L.J.; Kim, A.J.; Potter, S.J.; DeFalco, T.; Lincoln, J.; Yutzey, K.E. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 636–644. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, Q.; Liu, X.; Geng, B.; Dong, N.; Shi, J. Identifying Hub Genes of Calcific Aortic Valve Disease and Revealing the Immune Infiltration Landscape Based on Multiple WGCNA and Single-Cell Sequence Analysis. Front. Immunol. 2022, 13, 1035285. [Google Scholar] [CrossRef]
- Wu, L.-D.; Xiao, F.; Sun, J.-Y.; Li, F.; Chen, Y.-J.; Chen, J.-Y.; Zhang, J.; Qian, L.-L.; Wang, R.-X. Integrated Identification of Key Immune Related Genes and Patterns of Immune Infiltration in Calcified Aortic Valvular Disease: A Network Based Meta-Analysis. Front. Genet. 2022, 13, 971808. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Daoudi, M.; Rosa, M.; Vinod, M.; Louvet, L.; Copin, C.; Fanchon, M.; Vanhoutte, J.; Derudas, B.; Belloy, L.; et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ. Res. 2017, 121, 19–30. [Google Scholar] [CrossRef]
- Bartoli-Leonard, F.; Zimmer, J.; Aikawa, E. Innate and Adaptive Immunity: The Understudied Driving Force of Heart Valve Disease. Cardiovasc. Res. 2021, 117, 2506–2524. [Google Scholar] [CrossRef] [PubMed]
- Thériault, S.; Dina, C.; Messika-Zeitoun, D.; Le Scouarnec, S.; Capoulade, R.; Gaudreault, N.; Rigade, S.; Li, Z.; Simonet, F.; Lamontagne, M.; et al. Genetic Association Analyses Highlight IL6, ALPL, and NAV1 As 3 New Susceptibility Genes Underlying Calcific Aortic Valve Stenosis. Circ. Genom. Precis. Med. 2019, 12, e002617. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, W.; Ma, Z.; Li, L.; Chen, X. M1/M2 Macrophages and Associated Mechanisms in Congenital Bicuspid Aortic Valve Stenosis. Exp. Ther. Med. 2014, 7, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage Polarity in Cancer: A Review. J. Cell Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus Biofilms Prevent Macrophage Phagocytosis and Attenuate Inflammation In Vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef]
- Ushio, A.; Arakaki, R.; Yamada, A.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Crucial Roles of Macrophages in the Pathogenesis of Autoimmune Disease. World J. Immunol. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte Subsets Differentially Employ CCR2, CCR5, and CX3CR1 to Accumulate within Atherosclerotic Plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef]
- Chawla, A. Control of Macrophage Activation and Function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef]
- Johnson, J.L.; Newby, A.C. Macrophage Heterogeneity in Atherosclerotic Plaques. Curr. Opin. Lipidol. 2009, 20, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.C.; Aikawa, E. Roles and Regulation of Extracellular Vesicles in Cardiovascular Mineral Metabolism. Front. Cardiovasc. Med. 2018, 5, 187. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, R.; Katsuki, S.; Travers, R.; Romero, D.C.; Becker-Greene, D.; Passos, L.S.A.; Higashi, H.; Blaser, M.C.; Sukhova, G.K.; Buttigieg, J.; et al. S100A9-RAGE Axis Accelerates Formation of Macrophage-Mediated Extracellular Vesicle Microcalcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1838–1853. [Google Scholar] [CrossRef] [PubMed]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef]
- Raddatz, M.A.; Huffstater, T.; Bersi, M.R.; Reinfeld, B.I.; Madden, M.Z.; Booton, S.E.; Rathmell, W.K.; Rathmell, J.C.; Lindman, B.R.; Madhur, M.S.; et al. Macrophages Promote Aortic Valve Cell Calcification and Alter STAT3 Splicing. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e153–e165. [Google Scholar] [CrossRef]
- Tu, P.; Xu, Q.; Zhou, X.; Villa-Roel, N.; Kumar, S.; Dong, N.; Jo, H.; Ou, C.; Lin, Z. Myeloid CCN3 Protects against Aortic Valve Calcification. Cell Commun. Signal. 2023, 21, 14. [Google Scholar] [CrossRef]
- Li, G.; Qiao, W.; Zhang, W.; Li, F.; Shi, J.; Dong, N. The Shift of Macrophages toward M1 Phenotype Promotes Aortic Valvular Calcification. J. Thorac. Cardiovasc. Surg. 2017, 153, 1318–1327.e1. [Google Scholar] [CrossRef]
- Kaden, J.J.; Dempfle, C.-E.; Grobholz, R.; Tran, H.-T.; Kılıç, R.; Sarıkoç, A.; Brueckmann, M.; Vahl, C.; Hagl, S.; Haase, K.K.; et al. Interleukin-1 Beta Promotes Matrix Metalloproteinase Expression and Cell Proliferation in Calcific Aortic Valve Stenosis. Atherosclerosis 2003, 170, 205–211. [Google Scholar] [CrossRef]
- Weiss, R.M.; Miller, J.D.; Heistad, D.D. Fibrocalcific Aortic Valve Disease. Circ. Res. 2013, 113, 209–222. [Google Scholar] [CrossRef]
- Kaden, J.J.; Kiliç, R.; Sarikoç, A.; Hagl, S.; Lang, S.; Hoffmann, U.; Brueckmann, M.; Borggrefe, M. Tumor Necrosis Factor Alpha Promotes an Osteoblast-like Phenotype in Human Aortic Valve Myofibroblasts: A Potential Regulatory Mechanism of Valvular Calcification. Int. J. Mol. Med. 2005, 16, 869–872. [Google Scholar] [CrossRef]
- Khan, R.; Sheppard, R. Fibrosis in Heart Disease: Understanding the Role of Transforming Growth Factor-β 1 in Cardiomyopathy, Valvular Disease and Arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Grim, J.C.; Aguado, B.A.; Vogt, B.J.; Batan, D.; Andrichik, C.L.; Schroeder, M.E.; Gonzalez-Rodriguez, A.; Yavitt, F.M.; Weiss, R.M.; Anseth, K.S. Secreted Factors from Proinflammatory Macrophages Promote an Osteoblast-Like Phenotype in Valvular Interstitial Cells. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e296–e308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; The, E.; Nedumaran, B.; Ao, L.; Jarrett, M.J.; Xu, D.; Fullerton, D.A.; Meng, X. Monocytes Enhance the Inflammatory Response to TLR2 Stimulation in Aortic Valve Interstitial Cells through Paracrine Up-Regulation of TLR2 Level. Int. J. Biol. Sci. 2020, 16, 3062–3074. [Google Scholar] [CrossRef]
- Kakutani, Y.; Shioi, A.; Shoji, T.; Okazaki, H.; Koyama, H.; Emoto, M.; Inaba, M. Oncostatin M Promotes Osteoblastic Differentiation of Human Vascular Smooth Muscle Cells Through JAK3-STAT3 Pathway. J. Cell Biochem. 2015, 116, 1325–1333. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Shi, J.; Chen, S.; Xu, L.; Li, F.; Dong, N. IL-21 Promotes Osteoblastic Differentiation of Human Valvular Interstitial Cells through the JAK3/STAT3 Pathway. Int. J. Med. Sci. 2020, 17, 3065–3072. [Google Scholar] [CrossRef]
- Liu, J.; Sukhova, G.K.; Sun, J.-S.; Xu, W.-H.; Libby, P.; Shi, G.-P. Lysosomal Cysteine Proteases in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1359–1366. [Google Scholar] [CrossRef]
- Aikawa, E.; Aikawa, M.; Libby, P.; Figueiredo, J.-L.; Rusanescu, G.; Iwamoto, Y.; Fukuda, D.; Kohler, R.H.; Shi, G.-P.; Jaffer, F.A.; et al. Arterial and Aortic Valve Calcification Abolished by Elastolytic Cathepsin S Deficiency in Chronic Renal Disease. Circulation 2009, 119, 1785–1794. [Google Scholar] [CrossRef]
- Yip, C.Y.Y.; Chen, J.-H.; Zhao, R.; Simmons, C.A. Calcification by Valve Interstitial Cells Is Regulated by the Stiffness of the Extracellular Matrix. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 936–942. [Google Scholar] [CrossRef]
- Farrar, E.J.; Huntley, G.D.; Butcher, J. Endothelial-Derived Oxidative Stress Drives Myofibroblastic Activation and Calcification of the Aortic Valve. PLoS ONE 2015, 10, e0123257. [Google Scholar] [CrossRef]
- Mahler, G.J.; Farrar, E.J.; Butcher, J.T. Inflammatory Cytokines Promote Mesenchymal Transformation in Embryonic and Adult Valve Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Farrar, E.J.; Butcher, J.T. Heterogeneous Susceptibility of Valve Endothelial Cells to Mesenchymal Transformation in Response to TNFα. Ann. Biomed. Eng. 2014, 42, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Pabois, A.; Pagie, S.; Gérard, N.; Laboisse, C.; Pattier, S.; Hulin, P.; Nedellec, S.; Toquet, C.; Charreau, B. Notch Signaling Mediates Crosstalk between Endothelial Cells and Macrophages via Dll4 and IL6 in Cardiac Microvascular Inflammation. Biochem. Pharmacol. 2016, 104, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, E.; Pérez, M.A.; Rubio, A.; Ruiz-Hidalgo, M.J.; Baladrón, V.; García-Ramírez, J.J.; Gómez, J.C.; Laborda, J.; Diíaz-Guerra, M.J.M. Notch-1 Up-Regulation and Signaling Following Macrophage Activation Modulates Gene Expression Patterns Known to Affect Antigen-Presenting Capacity and Cytotoxic Activity. J. Immunol. 2006, 176, 5362–5373. [Google Scholar] [CrossRef]
- Jönsson, J.-I.; Xiang, Z.; Pettersson, M.; Lardelli, M.; Nilsson, G. Distinct and Regulated Expression of Notch Receptors in Hematopoietic Lineages and during Myeloid Differentiation. Eur. J. Immunol. 2001, 31, 3240–3247. [Google Scholar] [CrossRef]
- Singh, N.; Phillips, R.A.; Iscove, N.N.; Egan, S.E. Expression of Notch Receptors, Notch Ligands, and Fringe Genes in Hematopoiesis. Exp. Hematol. 2000, 28, 527–534. [Google Scholar] [CrossRef]
- Van der Wal, A.C.; Dingemans, K.P.; van den Bergh Weerman, M.; Das, P.K.; Becker, A.E. Specialized Membrane Contacts between Immunocompetent Cells in Human Atherosclerotic Plaques. Cardiovasc. Pathol. 1994, 3, 81–85. [Google Scholar] [CrossRef]
- Fung, E.; Tang, S.-M.T.; Canner, J.P.; Morishige, K.; Arboleda-Velasquez, J.F.; Cardoso, A.A.; Carlesso, N.; Aster, J.C.; Aikawa, M. Delta-Like 4 Induces Notch Signaling in Macrophages. Circulation 2007, 115, 2948–2956. [Google Scholar] [CrossRef]
- Keewan, E.; Naser, S.A. The Role of Notch Signaling in Macrophages during Inflammation and Infection: Implication in Rheumatoid Arthritis? Cells 2020, 9, 111. [Google Scholar] [CrossRef]
- Guo, S.; Liu, J.; Zhang, Y.; Liang, S.; Jiang, X.; Lin, Y. Notch Signaling Regulates M1-Type Polarization in Macrophages by Inhibiting Signal Regulatory Protein α (SIRPα). Chin. J. Cell. Mol. Immunol. 2021, 37, 673–678. [Google Scholar]
- Li, Z.; Nie, M.; Yu, L.; Tao, D.; Wang, Q.; He, Y.; Liu, Y.; Zhang, Y.; Han, H.; Wang, H. Blockade of the Notch Signaling Pathway Promotes M2 Macrophage Polarization to Suppress Cardiac Fibrosis Remodeling in Mice with Myocardial Infarction. Front. Cardiovasc. Med. 2022, 8, 639476. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; He, F.; Feng, F.; Liu, X.-W.; Dong, G.-Y.; Qin, H.-Y.; Hu, X.-B.; Zheng, M.-H.; Liang, L.; Feng, L.; et al. Notch Signaling Determines the M1 versus M2 Polarization of Macrophages in Antitumor Immune Responses. Cancer Res. 2010, 70, 4840–4849. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, Q.; Su, S.; Dong, W.; Zong, S.; Ma, Q.; Yang, X.; Zuo, D.; Zheng, S.; Meng, X.; et al. Interleukin 37 Suppresses M1 Macrophage Polarization Through Inhibition of the Notch1 and Nuclear Factor Kappa B Pathways. Front. Cell Dev. Biol. 2020, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Dayawansa, N.H.; Baratchi, S.; Peter, K. Uncoupling the Vicious Cycle of Mechanical Stress and Inflammation in Calcific Aortic Valve Disease. Front. Cardiovasc. Med. 2022, 9, 783543. [Google Scholar] [CrossRef] [PubMed]
- Foldi, J.; Chung, A.Y.; Xu, H.; Zhu, J.; Outtz, H.H.; Kitajewski, J.; Li, Y.; Hu, X.; Ivashkiv, L.B. Autoamplification of Notch Signaling in Macrophages by TLR-Induced and RBP-J–Dependent Induction of Jagged1. J. Immunol. 2010, 185, 5023–5031. [Google Scholar] [CrossRef]
- Palaga, T.; Buranaruk, C.; Rengpipat, S.; Fauq, A.H.; Golde, T.E.; Kaufmann, S.H.E.; Osborne, B.A. Notch Signaling Is Activated by TLR Stimulation and Regulates Macrophage Functions. Eur. J. Immunol. 2008, 38, 174–183. [Google Scholar] [CrossRef]
- Hu, X.; Chung, A.Y.; Wu, I.; Foldi, J.; Chen, J.; Ji, J.D.; Tateya, T.; Kang, Y.J.; Han, J.; Gessler, M.; et al. Integrated Regulation of Toll-like Receptor Responses by Notch and Interferon-γ Pathways. Immunity 2008, 29, 691–703. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, N.; Kim, M.; Woo, S.-H.; Han, I.; Park, J.; Kim, K.; Park, K.S.; Kim, K.; Shim, D.; et al. Single-Cell Transcriptomics Reveal Cellular Diversity of Aortic Valve and the Immunomodulation by PPARγ during Hyperlipidemia. Nat. Commun. 2022, 13, 5461. [Google Scholar] [CrossRef]
- Liu, W.; Yin, Y.; Zhou, Z.; He, M.; Dai, Y. OxLDL-Induced IL-1beta Secretion Promoting Foam Cells Formation Was Mainly via CD36 Mediated ROS Production Leading to NLRP3 Inflammasome Activation. Inflamm. Res. 2014, 63, 33–43. [Google Scholar] [CrossRef]
- Jovinge, S.; Ares, M.P.S.; Kallin, B.; Nilsson, J. Human Monocytes/Macrophages Release TNF-α in Response to Ox-LDL. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1573–1579. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.B.; van der Meer, J.W.M.; Netea, M.G.; Riksen, N.P. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, P.; Steinmetz, M.; Ackerschott, A.; Dregger, H.; Jehle, J.; Nickenig, G.; Latz, E.; Zimmer, S. P5128 Role of CLEC4E Expression in Development of Aortic Valve Stenosis. Eur. Heart J. 2018, 39, ehy566–P5128. [Google Scholar] [CrossRef]
- Clément, M.; Basatemur, G.; Masters, L.; Baker, L.; Bruneval, P.; Iwawaki, T.; Kneilling, M.; Yamasaki, S.; Goodall, J.; Mallat, Z. Necrotic Cell Sensor Clec4e Promotes a Proatherogenic Macrophage Phenotype Through Activation of the Unfolded Protein Response. Circulation 2016, 134, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lei, H.; Liu, L.; Xu, D. Lipoprotein(a), a Lethal Player in Calcific Aortic Valve Disease. Front. Cell Dev. Biol. 2022, 10, 812368. [Google Scholar] [CrossRef]
- Schlotter, F.; de Freitas, R.C.C.; Rogers, M.A.; Blaser, M.C.; Wu, P.-J.; Higashi, H.; Halu, A.; Iqbal, F.; Andraski, A.B.; Rodia, C.N.; et al. ApoC-III Is a Novel Inducer of Calcification in Human Aortic Valves. J. Biol. Chem. 2021, 296, 100193. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035. [Google Scholar] [CrossRef]
- Weinberg, J.B.; Misukonis, M.A.; Shami, P.J.; Mason, S.N.; Sauls, D.L.; Dittman, W.A.; Wood, E.R.; Smith, G.K.; McDonald, B.; Bachus, K.E. Human Mononuclear Phagocyte Inducible Nitric Oxide Synthase (INOS): Analysis of INOS MRNA, INOS Protein, Biopterin, and Nitric Oxide Production by Blood Monocytes and Peritoneal Macrophages. Blood 1995, 86, 1184–1195. [Google Scholar] [CrossRef]
- Cros, J.; Cagnard, N.; Woollard, K.; Patey, N.; Zhang, S.-Y.; Senechal, B.; Puel, A.; Biswas, S.K.; Moshous, D.; Picard, C.; et al. Human CD14dim Monocytes Patrol and Sense Nucleic Acids and Viruses via TLR7 and TLR8 Receptors. Immunity 2010, 33, 375–386. [Google Scholar] [CrossRef]
- Tapp, L.D.; Shantsila, E.; Wrigley, B.J.; Pamukcu, B.; Lip, G.Y.H. The CD14++CD16+ Monocyte Subset and Monocyte-platelet Interactions in Patients with ST-elevation Myocardial Infarction. J. Thromb. Haemost. 2012, 10, 1231–1241. [Google Scholar] [CrossRef]
- Barisione, C.; Garibaldi, S.; Ghigliotti, G.; Fabbi, P.; Altieri, P.; Casale, M.C.; Spallarossa, P.; Bertero, G.; Balbi, M.; Corsiglia, L.; et al. CD14CD16 Monocyte Subset Levels in Heart Failure Patients. Dis. Markers 2010, 28, 115–124. [Google Scholar] [CrossRef]
- Hewing, B.; Au, S.C.-D.; Ludwig, A.; Ellerbroek, R.; van Dijck, P.; Hartmann, L.; Grubitzsch, H.; Giannini, C.; Laule, M.; Stangl, V.; et al. Severe Aortic Valve Stenosis in Adults Is Associated with Increased Levels of Circulating Intermediate Monocytes. J. Cardiovasc. Transl. Res. 2017, 10, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Bartoli-Leonard, F.; Pennel, T.; Caputo, M. Immunotherapy in the Context of Aortic Valve Diseases. Cardiovasc. Drugs Ther. 2024, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Kraus, S.; Pierer, M.; Baerwald, C.; Wagner, U. The CD14 bright CD16+ Monocyte Subset Is Expanded in Rheumatoid Arthritis and Promotes Expansion of the Th17 Cell Population. Arthritis Rheum. 2012, 64, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Moniuszko, M.; Bodzenta-Lukaszyk, A.; Kowal, K.; Lenczewska, D.; Dabrowska, M. Enhanced Frequencies of CD14++CD16+, but Not CD14+CD16+, Peripheral Blood Monocytes in Severe Asthmatic Patients. Clin. Immunol. 2009, 130, 338–346. [Google Scholar] [CrossRef]
- Naicker, S.D.; Cormican, S.; Griffin, T.P.; Maretto, S.; Martin, W.P.; Ferguson, J.P.; Cotter, D.; Connaughton, E.P.; Dennedy, M.C.; Griffin, M.D. Chronic Kidney Disease Severity Is Associated with Selective Expansion of a Distinctive Intermediate Monocyte Subpopulation. Front. Immunol. 2018, 9, 2845. [Google Scholar] [CrossRef]
- Heine, G.H.; Ulrich, C.; Seibert, E.; Seiler, S.; Marell, J.; Reichart, B.; Krause, M.; Schlitt, A.; Köhler, H.; Girndt, M. CD14++CD16+ Monocytes but Not Total Monocyte Numbers Predict Cardiovascular Events in Dialysis Patients. Kidney Int. 2008, 73, 622–629. [Google Scholar] [CrossRef]
- Lo, S.-C.; Lee, W.-J.; Chen, C.-Y.; Lee, B.-C. Intermediate CD14++CD16+ Monocyte Predicts Severe Coronary Stenosis and Extensive Plaque Involvement in Asymptomatic Individuals. Int. J. Cardiovasc. Imaging 2017, 33, 1223–1236. [Google Scholar] [CrossRef]
- Hewing, B.; Ellerbroek, R.; Au, S.; Stangl, V.; Dreger, H.; Laule, M.; Grubitzsch, H.; Knebel, F.; Baumann, G.; Ludwig, A.; et al. Levels of Circulating Intermediate Monocytes Decrease after Aortic Valve Replacement in Patients with Severe Aortic Stenosis. Thromb. Haemost. 2017, 117, 2346–2355. [Google Scholar] [CrossRef]
- Neuser, J.; Galuppo, P.; Fraccarollo, D.; Willig, J.; Kempf, T.; Berliner, D.; Bauersachs, J.; Widder, J.D. Intermediate CD14++CD16+ Monocytes Decline after Transcatheter Aortic Valve Replacement and Correlate with Functional Capacity and Left Ventricular Systolic Function. PLoS ONE 2017, 12, e0183670. [Google Scholar] [CrossRef]
- Broeders, W.; Bekkering, S.; El Messaoudi, S.; Joosten, L.A.B.; van Royen, N.; Riksen, N.P. Innate Immune Cells in the Pathophysiology of Calcific Aortic Valve Disease: Lessons to Be Learned from Atherosclerotic Cardiovascular Disease? Basic. Res. Cardiol. 2022, 117, 28. [Google Scholar] [CrossRef]
- Castro, R.C.; Zambuzi, F.A.; Fontanari, C.; de Morais, F.R.; Bollela, V.R.; Kunkel, S.L.; Schaller, M.A.; Frantz, F.G. NOTCH1 and DLL4 Are Involved in the Human Tuberculosis Progression and Immune Response Activation. Tuberculosis 2020, 124, 101980. [Google Scholar] [CrossRef] [PubMed]
- Abplanalp, W.T.; John, D.; Cremer, S.; Assmus, B.; Dorsheimer, L.; Hoffmann, J.; Becker-Pergola, G.; Rieger, M.A.; Zeiher, A.M.; Vasa-Nicotera, M.; et al. Single-Cell RNA-Sequencing Reveals Profound Changes in Circulating Immune Cells in Patients with Heart Failure. Cardiovasc. Res. 2021, 117, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K.; Schnorr, P.J.; Pang, W.W.; Chao, M.P.; Chhabra, A.; Seita, J.; Feng, M.; Weissman, I.L. Myeloid Cell Origins, Differentiation, and Clinical Implications. Microbiol. Spectr. 2016, 4, 857–875. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Lee, W.-S.; Kim, J. Therapeutic Strategy for Atherosclerosis Based on Bone-Vascular Axis Hypothesis. Pharmacol. Ther. 2020, 206, 107436. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The Immunology of Atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial Infarction Accelerates Atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef]
- Heuser, M.; Thol, F.; Ganser, A. Clonal Hematopoiesis of Indeterminate Potential. Dtsch. Arztebl. Int. 2016, 113, 317. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Palumbo, D.; Fortini, F.; D’Agostino, Y.; Cimaglia, P.; Marracino, L.; Severi, P.; Strianese, O.; Tarallo, R.; Nassa, G.; et al. Transcriptomic Profiling of Calcified Aortic Valves in Clonal Hematopoiesis of Indeterminate Potential Carriers. Sci. Rep. 2022, 12, 20400. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal Haematopoiesis in Patients with Degenerative Aortic Valve Stenosis Undergoing Transcatheter Aortic Valve Implantation. Eur. Heart J. 2020, 41, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal Hematopoiesis Associated with TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Domínguez-Andrés, J.; Joosten, L.A.B.; Riksen, N.P.; Netea, M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021, 39, 667–693. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, C.D.C.C.; Groh, L.; Keating, S.T.; Kaffa, C.; Noz, M.P.; Kersten, S.; van Herwaarden, A.E.; Hoischen, A.; Joosten, L.A.B.; Timmers, H.J.L.M.; et al. Catecholamines Induce Trained Immunity in Monocytes In Vitro and In Vivo. Circ. Res. 2020, 127, 269–283. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Noz, M.P.; Bekkering, S.; Groh, L.; Nielen, T.M.; Lamfers, E.J.; Schlitzer, A.; El Messaoudi, S.; van Royen, N.; Huys, E.H.; Preijers, F.W.; et al. Reprogramming of Bone Marrow Myeloid Progenitor Cells in Patients with Severe Coronary Artery Disease. Elife 2020, 9, e60939. [Google Scholar] [CrossRef]
- Flores-Gomez, D.; Bekkering, S.; Netea, M.G.; Riksen, N.P. Trained Immunity in Atherosclerotic Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 62–69. [Google Scholar] [CrossRef]
- Bekkering, S.; Stiekema, L.C.A.; Bernelot Moens, S.; Verweij, S.L.; Novakovic, B.; Prange, K.; Versloot, M.; Roeters van Lennep, J.E.; Stunnenberg, H.; de Winther, M.; et al. Treatment with Statins Does Not Revert Trained Immunity in Patients with Familial Hypercholesterolemia. Cell Metab. 2019, 30, 1–2. [Google Scholar] [CrossRef]
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.B.; Netea, M.G.; Gomes, M.E.R.; et al. Innate Immune Cell Activation and Epigenetic Remodeling in Symptomatic and Asymptomatic Atherosclerosis in Humans in Vivo. Atherosclerosis 2016, 254, 228–236. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klauzen, P.; Basovich, L.; Shishkova, D.; Markova, V.; Malashicheva, A. Macrophages in Calcific Aortic Valve Disease: Paracrine and Juxtacrine Disease Drivers. Biomolecules 2024, 14, 1547. https://doi.org/10.3390/biom14121547
Klauzen P, Basovich L, Shishkova D, Markova V, Malashicheva A. Macrophages in Calcific Aortic Valve Disease: Paracrine and Juxtacrine Disease Drivers. Biomolecules. 2024; 14(12):1547. https://doi.org/10.3390/biom14121547
Chicago/Turabian StyleKlauzen, Polina, Liubov Basovich, Daria Shishkova, Victoria Markova, and Anna Malashicheva. 2024. "Macrophages in Calcific Aortic Valve Disease: Paracrine and Juxtacrine Disease Drivers" Biomolecules 14, no. 12: 1547. https://doi.org/10.3390/biom14121547
APA StyleKlauzen, P., Basovich, L., Shishkova, D., Markova, V., & Malashicheva, A. (2024). Macrophages in Calcific Aortic Valve Disease: Paracrine and Juxtacrine Disease Drivers. Biomolecules, 14(12), 1547. https://doi.org/10.3390/biom14121547