
Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses
 , , and
, , and
Abstract
:1. Introduction
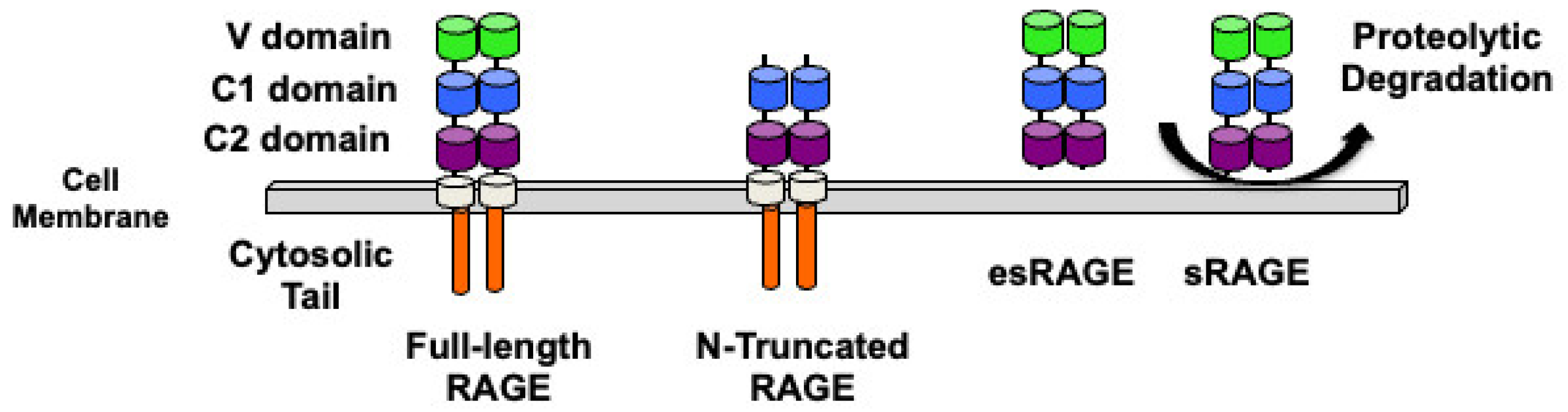
2. RAGE Structure and Isoforms
3. Signaling Pathways Activated Following RAGE Interaction with Its Ligands
4. RAGE Ligands and Their Effects on Immune Cells
4.1. Advanced Glycation End Products (AGE)
4.2. High-Mobility Group Box 1 (HMGB1) Protein
4.3. Nucleic Acid
4.4. MAC-1
4.5. C1q
4.6. Phosphatidylserine
4.7. Amyloid β Peptides
4.8. S100 Proteins
5. RAGE Expression in Immune Cells and Its Role in Immune Responses
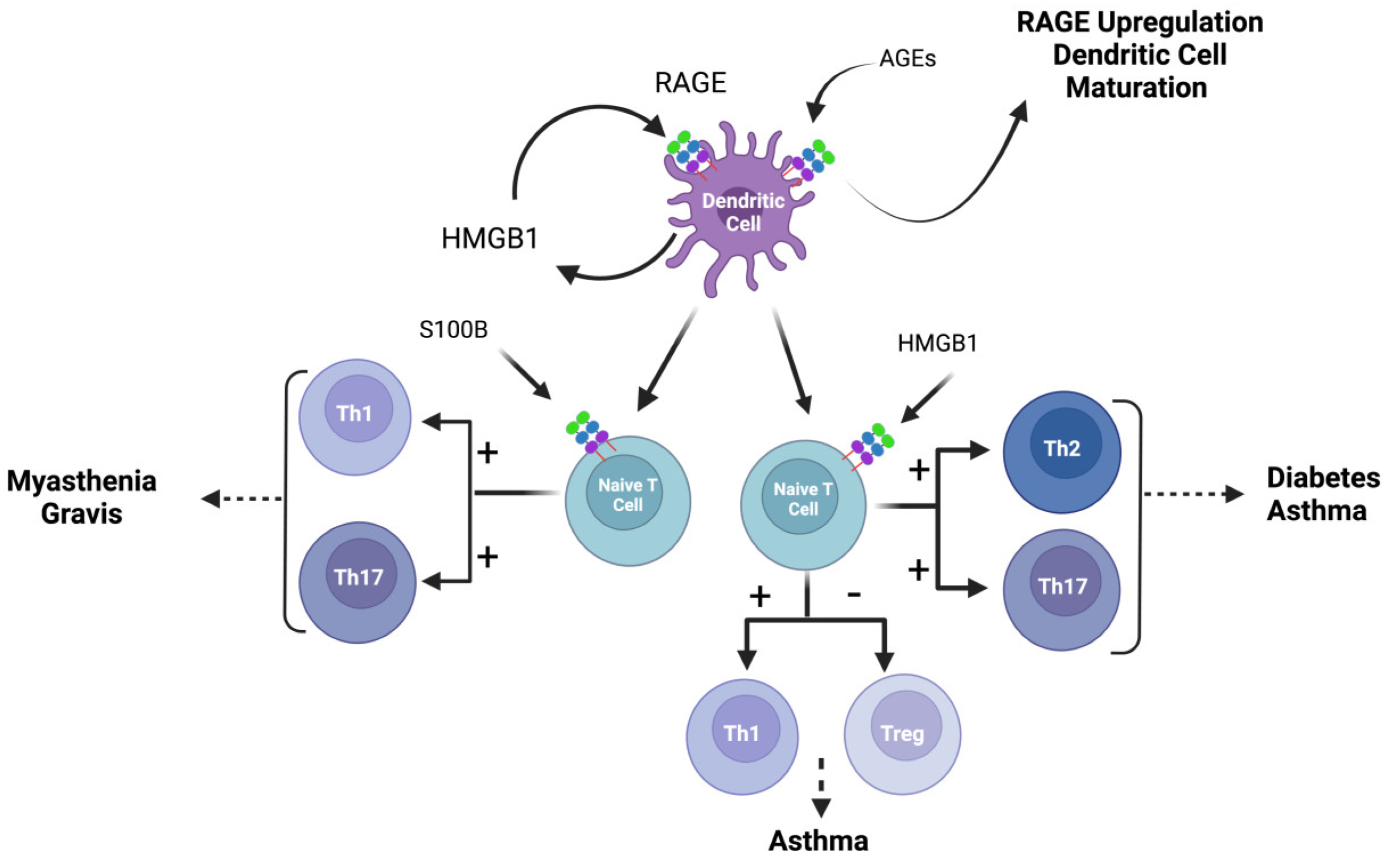
5.1. RAGE in Dendritic Cells
5.2. RAGE in T Cells
5.3. RAGE in Monocytes and Macrophages
5.3.1. RAGE and Macrophage Polarization in Diabetic Neuropathy
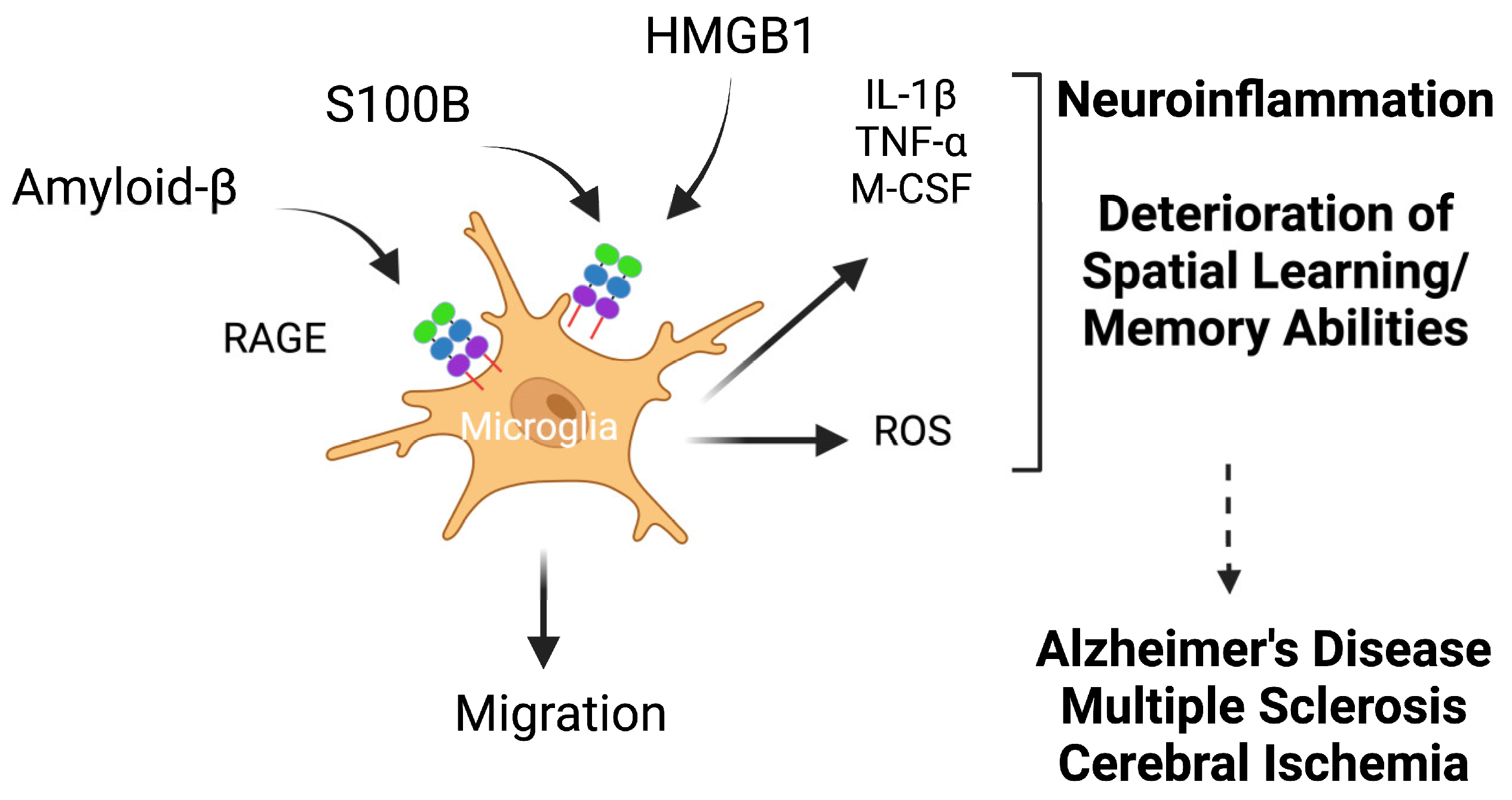
5.3.2. RAGE and Microglia in Alzheimer’s Disease
5.3.3. Macrophage RAGE in Pancreatic Tumor Microenvironment
5.3.4. RAGE and Macrophages in Adipose Tissues
5.3.5. RAGE in Sepsis
5.3.6. RAGE and Cholesterol Efflux in Macrophages
5.3.7. Macrophage RAGE in Atherosclerotic Plaques
5.3.8. RAGE in Myocardial Fibrosis
5.3.9. RAGE in Lung Fibrosis
5.4. RAGE in Granulocytes
5.4.1. RAGE in Neutrophils
5.4.2. RAGE in Eosinophils and in Eosinophilic Asthma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the Distribution of a Newly Characterized Receptor for Advanced Glycation End Products in Tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar] [PubMed]
- Fehrenbach, H.; Kasper, M.; Tschernig, T.; Shearman, M.S.; Schuh, D.; Muller, M. Receptor for Advanced Glycation Endproducts (RAGE) Exhibits Highly Differential Cellular and Subcellular Localisation in Rat and Human Lung. Cell. Mol. Biol. 1998, 44, 1147–1157. [Google Scholar] [PubMed]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in Lung Disease: The Receptor for Advanced Glycation Endproducts (RAGE) is a Major Mediator of Pulmonary Inflammatory Responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Hofmann, M.; Taguchi, A.; Yan, S.D.; Stern, D.M. RAGE: A Multiligand Receptor Contributing to the Cellular Response in Diabetic Vasculopathy and Inflammation. Semin. Thromb. Hemost. 2000, 26, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the Receptor for Advanced Glycation End Products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The Biology of the Receptor for Advanced Glycation End Products and Its Ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Donato, R. RAGE: A Single Receptor for Several Ligands and Different Cellular Responses: The Case of Certain S100 Proteins. Curr. Mol. Med. 2007, 7, 711–724. [Google Scholar] [CrossRef]
- Fritz, G. RAGE: A single Receptor Fits Multiple Ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Alexiou, P.; Chatzopoulou, M.; Pegklidou, K.; Demopoulos, V.J. RAGE: A Multi-Ligand Receptor Unveiling Novel Insights in Health and Disease. Curr. Med. Chem. 2010, 17, 2232–2252. [Google Scholar] [CrossRef]
- Bucciarelli, L.G.; Wendt, T.; Rong, L.; Lalla, E.; Hofmann, M.A.; Goova, M.T.; Taguchi, A.; Yan, S.F.; Yan, S.D.; Stern, D.M.; et al. RAGE is a Multiligand Receptor of the Immunoglobulin Superfamily: Implications for Homeostasis and Chronic Disease. Cell Mol. Life Sci. 2002, 59, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Du Yan, S.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; et al. Amyloid-Beta Peptide-Receptor for Advanced Glycation Endproduct Interaction Elicits Neuronal Expression of Macrophage-Colony Stimulating Factor: A Proinflammatory Pathway in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar] [CrossRef] [PubMed]
- Sturchler, E.; Galichet, A.; Weibel, M.; Leclerc, E.; Heizmann, C.W. Site-Specific Blockade of RAGE-Vd Prevents Amyloid-Beta Oligomer Neurotoxicity. J. Neurosci. 2008, 28, 5149–5158. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE Binds C1q and Enhances C1q-Mediated Phagocytosis. Cell Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Toure, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J.; et al. Lysophosphatidic Acid Targets Vascular and Oncogenic Pathways Via RAGE Signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE Binds Preamyloid IAPP Intermediates and Mediates Pancreatic Beta Cell Proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE is a Nucleic Acid Receptor that Promotes Inflammatory Responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and Expression of A Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, Classification, and Expression of RAGE Gene Splice Variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef]
- Bierhaus, A.; Nawroth, P.P. Multiple Levels of Regulation Determine the Role of the Receptor for AGE (RAGE) As Common Soil in Inflammation, Immune Responses and Diabetes Mellitus and its Complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef]
- Nickel, W.; Rabouille, C. Mechanisms of Regulated Unconventional Protein Secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Grone, H.J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for Advanced Glycation end Products (RAGE) Regulates Sepsis but Not the Adaptive Immune Response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yan, S.S.; Colgan, J.; Zhang, H.P.; Luban, J.; Schmidt, A.M.; Stern, D.; Herold, K.C. Blockade of Late Stages of Autoimmune Diabetes by Inhibition of the Receptor for Advanced Glycation End Products. J. Immunol. 2004, 173, 1399–1405. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 Proteins to RAGE: An Update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE Cytoplasmic Domain with Diaphanous-1 Is Required for Ligand-Stimulated Cellular Migration through Activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.H. TIRAP, An Adaptor Protein for TLR2/4, Transduces a Signal from RAGE Phosphorylated Upon Ligand Binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef]
- Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. The RAGE/DIAPH1 Signaling Axis & Implications for the Pathogenesis of Diabetic Complications. Int. J. Mol. Sci. 2022, 23, 4579. [Google Scholar] [CrossRef]
- Malherbe, P.; Richards, J.G.; Gaillard, H.; Thompson, A.; Diener, C.; Schuler, A.; Huber, G. cDNA Cloning of a Novel Secreted Isoform of the Human Receptor for Advanced Glycation End Products and Characterization of Cells Co-Expressing Cell-Surface Scavenger Receptors and Swedish Mutant Amyloid Precursor Protein. Brain Res. Mol. Brain Res. 1999, 71, 159–170. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel Splice Variants of the Receptor for Advanced Glycation End-Products Expressed in Human Vascular Endothelial Cells and Pericytes, and their Putative Roles in Diabetes-Induced Vascular Injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Ding, Q.; Keller, J.N. Evaluation of RAGE Isoforms, Ligands, and Signaling in the Brain. Biochim. Biophys. Acta 2005, 1746, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Galichet, A.; Weibel, M.; Heizmann, C.W. Calcium-Regulated Intramembrane Proteolysis of the RAGE Receptor. Biochem. Biophys. Res. Commun. 2008, 370, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A Soluble form of the Receptor for Advanced Glycation Endproducts (RAGE) Is Produced by Proteolytic Cleavage of the Membrane-Bound form by the Sheddase a Disintegrin and Metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Reiniger, N.; Yang, H.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternative Splicing of the Murine Receptor for Advanced Glycation End-Products (RAGE) Gene. FASEB J. 2009, 23, 1766–1774. [Google Scholar] [CrossRef]
- Maillard-Lefebvre, H.; Boulanger, E.; Daroux, M.; Gaxatte, C.; Hudson, B.I.; Lambert, M. Soluble Receptor for Advanced Glycation End Products: A New Biomarker in Diagnosis and Prognosis of Chronic Inflammatory Diseases. Rheumatology 2009, 48, 1190–1196. [Google Scholar] [CrossRef]
- Vazzana, N.; Santilli, F.; Cuccurullo, C.; Davi, G. Soluble Forms of RAGE in Internal Medicine. Intern. Emerg. Med. 2009, 4, 389–401. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T.; Nakamura, K. Kinetics, Role and Therapeutic Implications of Endogenous Soluble Form of Receptor for Advanced Glycation End Products (sRAGE) in Diabetes. Curr. Drug Targets 2007, 8, 1138–1143. [Google Scholar] [CrossRef]
- Schmidt, A.M. Soluble RAGEs–Prospects for Treating & Tracking Metabolic and Inflammatory Disease. Vasc. Pharmacol. 2015, 72, 389–401. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The Multiligand Receptor RAGE as a Progression Factor Amplifying Immune and Inflammatory Responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Yan, S.F.; Du Yan, S.; Ramasamy, R.; Schmidt, A.M. Tempering the Wrath of RAGE: An Emerging Therapeutic Strategy Against Diabetic Complications, Neurodegeneration, and Inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The Expression of the Receptor for Advanced Glycation Endproducts (RAGE) is Permissive for Early Pancreatic Neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of Receptor for Advanced Glycation End-Product (RAGE) and the JAK/STAT-Signaling Pathway in AGE-Induced Collagen Production in NRK-49F Cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Riehl, A.; Nemeth, J.; Angel, P.; Hess, J. The Receptor RAGE: Bridging Inflammation and Cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Schmidt, A.M. Characterization and Functional Analysis of the Promoter of RAGE, the Receptor for Advanced Glycation End Products. J. Biol. Chem. 1997, 272, 16498–16506. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Rauvala, H. Receptor for Advanced Glycation End Products (RAGE) Signaling Induces CREB-Dependent Chromogranin Expression During Neuronal Differentiation. J. Biol. Chem. 2002, 277, 38635–38646. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-Associated Sustained Activation of the Transcription Factor Nuclear Factor-kappab. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Nawroth, P.P. RAGE (Receptor for Advanced Glycation End Products): A Central Player in the Inflammatory Response. Microbes Infect. 2004, 6, 1219–1225. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Naka, Y.; Schmidt, A.M. Glycation, Inflammation, and RAGE: A Scaffold for the Macrovascular Complications of Diabetes and Beyond. Circ. Res. 2003, 93, 1159–1169. [Google Scholar] [CrossRef]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J., Jr.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for Advanced Glycation End Products Mediates Inflammation and Enhanced Expression of Tissue Factor in Vasculature of Diabetic Apolipoprotein E-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and Their Role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for Advanced Glycation End Products-Binding COOH-Terminal Motif of Amphoterin Inhibits Invasive Migration and Metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar]
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of High Mobility Group Box 1 by Dendritic Cells Controls T Cell Activation Via the Receptor for Advanced Glycation End Products. J. Immunol. 2005, 174, 7506–7515. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, J.; Zhu, F.; Li, R.; Liu, B.; Xu, W.; He, G.; Cao, H.; Wang, Y.; Yang, J. HMGB1 Regulates T Helper 2 and T Helper17 Cell Differentiation Both Directly and Indirectly in Asthmatic Mice. Mol. Immunol. 2018, 97, 45–55. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Zhang, C.; Wang, Y.; Cui, W.; Li, H.; Du, J. S100a8/a9 Released by CD11b+Gr1+ Neutrophils Activates Cardiac Fibroblasts to Initiate Angiotensin II-Induced Cardiac Inflammation and Injury. Hypertension 2014, 63, 1241–1250. [Google Scholar] [CrossRef]
- Kang, J.H.; Hwang, S.M.; Chung, I.Y. S100A8, S100A9 and S100A12 Activate Airway Epithelial Cells to Produce MUC5AC Via Extracellular Signal-Regulated Kinase and Nuclear Factor-Kappab Pathways. Immunology 2015, 144, 79–90. [Google Scholar] [CrossRef]
- He, R.; Chen, Y.; Chen, X.; Yuan, B. Mechanism of miR-181a-5p in Regulatory T/T-Helper 17 Immune Imbalance and Asthma Development in Mice with Allergic Rhinitis. Int. Arch. Allergy Immunol. 2022, 183, 375–388. [Google Scholar] [CrossRef]
- He, C.; Sun, S.; Zhang, Y.; Xie, F.; Li, S. The Role of Irreversible Electroporation in Promoting M1 Macrophage Polarization Via Regulating the HMGB1-RAGE-MAPK Axis in Pancreatic Cancer. Oncoimmunology 2021, 10, 1897295. [Google Scholar] [CrossRef]
- Killian, K.N.; Kosanovich, J.L.; Lipp, M.A.; Empey, K.M.; Oury, T.D.; Perkins, T.N. RAGE Contributes to Allergen Driven Severe Neutrophilic Airway Inflammation Via NLRP3 Inflammasome Activation in Mice. Front. Immunol. 2023, 14, 1039997. [Google Scholar] [CrossRef]
- Ji, Z.; He, L.; Rotem, A.; Janzer, A.; Cheng, C.S.; Regev, A.; Struhl, K. Genome-Scale Identification of Transcription Factors that Mediate an Inflammatory Network During Breast Cellular Transformation. Nat. Commun. 2018, 9, 2068. [Google Scholar] [CrossRef]
- Miyata, T.; Hori, O.; Zhang, J.; Yan, S.D.; Ferran, L.; Iida, Y.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) Is a Central Mediator of the Interaction of AGE-beta2microglobulin with Human Mononuclear Phagocytes Via an Oxidant-Sensitive Pathway. Implications for the Pathogenesis of Dialysis-Related Amyloidosis. J. Clin. Investig. 1996, 98, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Hilmenyuk, T.; Bellinghausen, I.; Heydenreich, B.; Ilchmann, A.; Toda, M.; Grabbe, S.; Saloga, J. Effects of Glycation of the Model Food Allergen Ovalbumin on Antigen Uptake and Presentation by Human Dendritic Cells. Immunology 2010, 129, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Jia, Q.; Liang, C.; Luo, Y.; Huang, D.; Sun, A.; Wang, K.; Zou, Y.; Chen, H. Advanced Glycosylation End Products Might Promote Atherosclerosis through Inducing the Immune Maturation of Dendritic Cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Sick, E.; Brehin, S.; Andre, P.; Coupin, G.; Landry, Y.; Takeda, K.; Gies, J.P. Advanced Glycation End Products (AGEs) Activate Mast Cells. Br. J. Pharmacol. 2010, 161, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.H.; Dai, S.M.; Tang, G.S.; Zhang, J.; Ren, D.; Wang, Z.W.; Shen, Q. HMGB1 Enhances the Proinflammatory Activity of Lipopolysaccharide by Promoting the Phosphorylation of MAPK p38 through Receptor for Advanced Glycation End Products. J. Immunol. 2009, 183, 6244–6250. [Google Scholar] [CrossRef]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-Like Receptor 9-Dependent Activation by DNA-Containing Immune Complexes is Mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The Receptor for Advanced Glycation End Products (RAGE) Is a Cellular Binding Site for Amphoterin. Mediation of Neurite Outgrowth and Co-Expression of Rage and Amphoterin in the Developing Nervous System. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, Y.; Wang, J.; Shi, X.; Liu, Q.; Liu, Z.; Li, Y.; Scott, M.J.; Xiao, G.; Li, S.; et al. Macrophage Endocytosis of High-Mobility Group Box 1 Triggers Pyroptosis. Cell Death Differ. 2014, 21, 1229–1239. [Google Scholar] [CrossRef]
- Youn, J.H.; Oh, Y.J.; Kim, E.S.; Choi, J.E.; Shin, J.S. High Mobility Group Box 1 Protein Binding to Lipopolysaccharide Facilitates Transfer of Lipopolysaccharide to Cd14 and Enhances Lipopolysaccharide-Mediated TNF-Alpha Production in Human Monocytes. J. Immunol. 2008, 180, 5067–5074. [Google Scholar] [CrossRef]
- Deng, X.; Sun, L.; Lai, X.; Xiang, L.; Li, Q.; Zhang, W.; Zhang, L.; Sun, S. Tea Polypeptide Ameliorates Diabetic Nephropathy through RAGE and NF-κB Signaling Pathway in Type 2 Diabetes Mice. J. Agric. Food Chem. 2018, 66, 11957–11967. [Google Scholar] [CrossRef]
- Liu, L.; Yang, M.; Kang, R.; Dai, Y.; Yu, Y.; Gao, F.; Wang, H.; Sun, X.; Li, X.; Li, J.; et al. HMGB1-DNA Complex-Induced Autophagy Limits AIM2 Inflammasome Activation through RAGE. Biochem. Biophys. Res. Commun. 2014, 450, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fu, S.; Fan, X.G.; Lotze, M.T.; Zeh, H.J., 3rd; Tang, D.; Kang, R. Nuclear DAMP Complex-Mediated RAGE-Dependent Macrophage Cell Death. Biochem. Biophys. Res. Commun. 2015, 458, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Porat, A.; He, M.; Suurmond, J.; Santiago-Schwarz, F.; Andersson, U.; Coleman, T.R.; Volpe, B.T.; Tracey, K.J.; Al-Abed, Y.; et al. C1q and HMGB1 Reciprocally Regulate Human Macrophage Polarization. Blood 2016, 128, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xiang, A.; Peng, T.; Doran, A.C.; Tracey, K.J.; Barnes, B.J.; Tabas, I.; Son, M.; Diamond, B. HMGB1-C1q Complexes Regulate Macrophage Function by Switching between Leukotriene and Specialized Proresolving Mediator Biosynthesis. Proc. Natl. Acad. Sci. USA 2019, 116, 23254–23263. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The Pattern Recognition Receptor (RAGE) Is a Counterreceptor for Leukocyte Integrins: A Novel Pathway for Inflammatory Cell Recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Orlova, V.V.; Choi, E.Y.; Xie, C.; Chavakis, E.; Bierhaus, A.; Ihanus, E.; Ballantyne, C.M.; Gahmberg, C.G.; Bianchi, M.E.; Nawroth, P.P.; et al. A Novel Pathway of HMGB1-Mediated Inflammatory Cell Recruitment That Requires Mac-1-Integrin. EMBO J. 2007, 26, 1129–1139. [Google Scholar] [CrossRef]
- Lue, L.F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of Microglial Receptor for Advanced Glycation Endproducts (RAGE) in Alzheimer’s Disease: Identification of a Cellular Activation Mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef]
- Fang, F.; Lue, L.F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Schmidt, A.M.; Chen, J.X.; et al. RAGE-Dependent Signaling in Microglia Contributes to Neuroinflammation, Abeta Accumulation, and Impaired Learning/Memory in a Mouse Model of Alzheimer’s Disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and Amyloid-Beta Peptide Neurotoxicity in Alzheimer’s Disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Li, Z.H.; Dulyaninova, N.G.; House, R.P.; Almo, S.C.; Bresnick, A.R. S100A4 Regulates Macrophage Chemotaxis. Mol. Biol. Cell 2010, 21, 2598–2610. [Google Scholar] [CrossRef]
- Cerezo, J.; Zuniga, J.; Requena, A.; Avila Ferrer, F.J.; Santoro, F. Erratum: Harmonic Models in Cartesian and Internal Coordinates to Simulate the Absorption Spectra of Carotenoids at Finite Temperatures. J. Chem. Theory Comput. 2014, 10, 3586–3587. [Google Scholar] [CrossRef]
- Zhou, L.J.; Peng, J.; Chen, M.; Yao, L.J.; Zou, W.H.; He, C.Y.; Peng, H.J. Toxoplasma Gondii SAG1 Targeting Host Cell S100A6 for Parasite Invasion and Host Immunity. iScience 2021, 24, 103514. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; He, H.; Kristine, M.S.; Guan, W.; Gao, J.; Wang, Z.; Hu, J.; Han, L.; Li, J.; Han, W.; et al. Therapeutic Effects of Recombinant Human S100A6 and Soluble Receptor for Advanced Glycation End Products(sRAGE) on CCl(4)-Induced Liver Fibrosis in Mice. Eur. J. Pharmacol. 2018, 833, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Frosch, M.; Sorg, C.; Roth, J. Phagocyte-Specific Calcium-Binding S100 Proteins as Clinical Laboratory Markers of Inflammation. Clin. Chim. Acta 2004, 344, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Kinoshita, R.; Tomonobu, N.; Gohara, Y.; Tomida, S.; Takahashi, Y.; Senoo, S.; Taniguchi, A.; Itano, J.; Yamamoto, K.I.; et al. The Heterodimer S100A8/A9 Is a Potent Therapeutic Target for Idiopathic Pulmonary Fibrosis. J. Mol. Med. 2021, 99, 131–145. [Google Scholar] [CrossRef]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Makino, H. Increased Expression of Receptor for Advanced Glycation End Products by Synovial Tissue Macrophages in Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 97–104. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Y.; Shao, Y.; Lu, X.; Zhang, H.; Miao, C. S100A8/A9(hi) Neutrophils Induce Mitochondrial Dysfunction and PANoptosis in Endothelial Cells Via Mitochondrial Complex I Deficiency During Sepsis. Cell Death Dis. 2024, 15, 462. [Google Scholar] [CrossRef]
- Yang, Z.; Yan, W.X.; Cai, H.; Tedla, N.; Armishaw, C.; Di Girolamo, N.; Wang, H.W.; Hampartzoumian, T.; Simpson, J.L.; Gibson, P.G.; et al. S100A12 Provokes Mast Cell Activation: A Potential Amplification Pathway in Asthma and Innate Immunity. J. Allergy Clin. Immunol. 2007, 119, 106–114. [Google Scholar] [CrossRef]
- Adami, C.; Sorci, G.; Blasi, E.; Agneletti, A.L.; Bistoni, F.; Donato, R. S100B Expression in and Effects on Microglia. Glia 2001, 33, 131–142. [Google Scholar] [CrossRef]
- Hu, J.; Castets, F.; Guevara, J.L.; Van Eldik, L.J. S100 Beta Stimulates Inducible Nitric Oxide Synthase Activity and mRNA Levels in Rat Cortical Astrocytes. J. Biol. Chem. 1996, 271, 2543–2547. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of Neurite Outgrowth and Cell Survival by Amphoterin and S100 Proteins through Receptor for Advanced Glycation End Products (RAGE) Activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Lu, J.; Savani, C.; De Filippis, D.; Iuvone, T.; Steardo, L., Jr.; Sheen, V.; Steardo, L. S100B Induces Tau Protein Hyperphosphorylation Via Dickopff-1 Up-Regulation and Disrupts the Wnt Pathway in Human Neural Stem Cells. J. Cell Mol. Med. 2008, 12, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhu, W.; Zhang, Y.; Pan, S.; Bao, J. S100B Promotes Microglia M1 Polarization and Migration to Aggravate Cerebral Ischemia. Inflamm. Res. 2018, 67, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.T.; Tu, M.C.; Zhung, P. Advanced Glycation End Product (AGE): Characterization of the Products from the Reaction between D-Glucose and Serum Albumin. J. Clin. Lab. Anal. 1996, 10, 21–34. [Google Scholar] [CrossRef]
- Twarda-Clapa, A.; Olczak, A.; Bialkowska, A.M.; Koziolkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef]
- Vashishth, D.; Gibson, G.J.; Khoury, J.I.; Schaffler, M.B.; Kimura, J.; Fyhrie, D.P. Influence of Nonenzymatic Glycation on Biomechanical Properties of Cortical Bone. Bone 2001, 28, 195–201. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products: Sparking the Development of Diabetic Vascular Injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Sherwani, S.I.; Khan, H.A.; Ekhzaimy, A.; Masood, A.; Sakharkar, M.K. Significance of HbA1c Test in Diagnosis and Prognosis of Diabetic Patients. Biomark. Insights 2016, 11, 95–104. [Google Scholar] [CrossRef]
- Li, Y.M.; Mitsuhashi, T.; Wojciechowicz, D.; Shimizu, N.; Li, J.; Stitt, A.; He, C.; Banerjee, D.; Vlassara, H. Molecular Identity and Cellular Distribution of Advanced Glycation Endproduct Receptors: Relationship of P60 to OST-48 and P90 to 80K-H Membrane Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 11047–11052. [Google Scholar] [CrossRef]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Miyazaki, A.; Hakamata, H.; Horiuchi, S.; Nakayama, H. CD36, Serves as a Receptor for Advanced Glycation Endproducts (AGE). J. Diabetes Complicat. 2002, 16, 56–59. [Google Scholar] [CrossRef]
- el Khoury, J.; Thomas, C.A.; Loike, J.D.; Hickman, S.E.; Cao, L.; Silverstein, S.C. Macrophages Adhere to Glucose-Modified Basement Membrane Collagen Iv Via Their Scavenger Receptors. J. Biol. Chem. 1994, 269, 10197–10200. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Li, Y.M.; Imani, F.; Wojciechowicz, D.; Yang, Z.; Liu, F.T.; Cerami, A. Identification of Galectin-3 as a High-Affinity Binding Protein for Advanced Glycation End Products (AGE): A New Member of the AGE-Receptor Complex. Mol. Med. 1995, 1, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Vetter, S.W. Glycated Serum Albumin and AGE Receptors. Adv. Clin. Chem. 2015, 72, 205–275. [Google Scholar] [CrossRef]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced Glycation End-Products Produced Systemically and by Macrophages: A Common Contributor to Inflammation and Degenerative Diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Stern, D. Atherosclerosis and Diabetes: The RAGE Connection. Curr. Atheroscler. Rep. 2000, 2, 430–436. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Receptor for AGE (RAGE) and Its Ligands-Cast into Leading Roles in Diabetes and the Inflammatory Response. J. Mol. Med. 2009, 87, 235–247. [Google Scholar] [CrossRef]
- Yan, S.F.; Naka, Y.; Hudson, B.I.; Herold, K.; Yan, S.D.; Ramasamy, R.; Schmidt, A.M. The Ligand/RAGE Axis: Lighting the Fuse and Igniting Vascular Stress. Curr. Atheroscler. Rep. 2006, 8, 232–239. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, F.; Shi, H.; Gao, Y.; Dong, Z.; Ma, L.; Sun, X.; Li, X.; Chang, S.; Wang, Z.; et al. Neutrophil-Derived Advanced Glycation End Products-Nepsilon-(Carboxymethyl) Lysine Promotes RIP3-Mediated Myocardial Necroptosis Via RAGE and Exacerbates Myocardial Ischemia/Reperfusion Injury. FASEB J. 2019, 33, 14410–14422. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hasu, M.; Popov, D.; Zhang, J.H.; Chen, J.; Yan, S.D.; Brett, J.; Cao, R.; Kuwabara, K.; Costache, G.; et al. Receptor for Advanced Glycation End Products (AGEs) Has a Central Role in Vessel Wall Interactions and Gene Activation in Response to Circulating AGE Proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 8807–8811. [Google Scholar] [CrossRef]
- Sevillano, N.; Giron, M.D.; Salido, M.; Vargas, A.M.; Vilches, J.; Salto, R. Internalization of the Receptor for Advanced Glycation End Products (RAGE) is Required to Mediate Intracellular Responses. J. Biochem. 2009, 145, 21–30. [Google Scholar] [CrossRef]
- Schroter, D.; Hohn, A. Role of Advanced Glycation End Products in Carcinogenesis and Their Therapeutic Implications. Curr. Pharm. Des. 2018, 24, 5245–5251. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Glycolysis. Cold Spring Harb. Perspect. Biol. 2021, 13, a040535. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Eva, T.A.; Barua, N.; Chowdhury, M.M.; Yeasmin, S.; Rakib, A.; Islam, M.R.; Emran, T.B.; Simal-Gandara, J. Perspectives on Signaling for Biological-and Processed Food-Related Advanced Glycation End-Products and Its Role in Cancer Progression. Crit. Rev. Food Sci. Nutr. 2022, 62, 2655–2672. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A New Group of Chromatin-Associated Proteins with a High Content of Acidic and basic Amino Acids. Eur. J. Biochem. 1973, 38, 14–19. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in Health and Disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef]
- Tang, D.; Loze, M.T.; Zeh, H.J.; Kang, R. The Redox Protein HMGB1 Regulates Cell Death and Survival in Cancer Treatment. Autophagy 2010, 6, 1181–1183. [Google Scholar] [CrossRef]
- Willingham, S.B.; Allen, I.C.; Bergstralh, D.T.; Brickey, W.J.; Huang, M.T.; Taxman, D.J.; Duncan, J.A.; Ting, J.P. NLRP3 (NALP3, Cryopyrin) Facilitates in Vivo Caspase-1 Activation, Necrosis, and HMGB1 Release Via Inflammasome-Dependent and -Independent Pathways. J. Immunol. 2009, 183, 2008–2015. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Zeh, H.J., 3rd; Lotze, M.T. High-Mobility Group Box 1, Oxidative Stress, and Disease. Antioxid. Redox Signal 2011, 14, 1315–1335. [Google Scholar] [CrossRef]
- Andersson, U.; Yang, H.; Harris, H. High-mobility Group Box 1 Protein (HMGB1) Operates as an Alarmin Outside as Well as Inside Cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Falciola, L.; Ferrari, S.; Lilley, D.M. The DNA Binding Site of HMG1 Protein Is Composed of Two Similar Segments (HMG Boxes), Both of Which Have Counterparts in Other Eukaryotic Regulatory Proteins. EMBO J. 1992, 11, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The Many Faces of HMGB1: Molecular Structure-Functional Activity in Inflammation, Apoptosis, and Chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic Cells Hyperacetylate Chromatin Protein HMGB1 to Redirect It Towards Secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, P.M.; Doggett, T.A.; Choi, J.; Hancock, M.A.; Durocher, Y.; Frank, F.; Nagar, B.; Ferguson, T.A.; Saleh, M. An Immunogenic Peptide in the A-Box of HMGB1 Protein Reverses Apoptosis-Induced Tolerance through RAGE Receptor. J. Biol. Chem. 2014, 289, 7777–7786. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The Mechanism of HMGB1 Secretion and Release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The Nuclear Protein HMGB1 is Secreted by Monocytes Via a Non-Classical, Vesicle-Mediated Secretory Pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-Dependent Release of the Alarmin HMGB1 in Endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef]
- Bianchi, M.E. HMGB1 Loves Company. J. Leukoc. Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef]
- Abib, R.T.; Quincozes-Santos, A.; Nardin, P.; Wofchuk, S.T.; Perry, M.L.; Goncalves, C.A.; Gottfried, C. Epicatechin Gallate Increases Glutamate Uptake and S100B Secretion in C6 Cell Lineage. Mol. Cell Biochem. 2008, 310, 153–158. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Y.; Zhang, P.; Li, Y.; Yang, Y.; Yang, Y.; Zhu, J.; Song, X.; Jiang, G.; Fan, J. Hemorrhagic Shock Primes for Lung Vascular Endothelial Cell Pyroptosis: Role in Pulmonary Inflammation Following LPS. Cell Death Dis. 2016, 7, e2363. [Google Scholar] [CrossRef]
- Muller, S.; Bianchi, M.E.; Knapp, S. Thermodynamics of HMGB1 Interaction with Duplex DNA. Biochemistry 2001, 40, 10254–10261. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Ban, T.; Wang, Z.; Choi, M.K.; Kawamura, T.; Negishi, H.; Nakasato, M.; Lu, Y.; Hangai, S.; Koshiba, R.; et al. HMGB Proteins Function as Universal Sentinels for Nucleic-Acid-Mediated Innate Immune Responses. Nature 2009, 462, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A Novel Role for HMGB1 in TLR9-Mediated Inflammatory Responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Huang, Y.P.; Chou, C.H.; Ho, M.W.; Lin, H.J.; Chen, C.Y.; Wu, H.Y.; Lai, Y.R.; Lee, Y.H.; Chiu, C.H.; et al. RAGE Participates in the Intracellular Transport of Campylobacter Jejuni Cytolethal Distending Toxin. J. Microbiol. Immunol. Infect. 2024, 57, 709–719. [Google Scholar] [CrossRef]
- Bertheloot, D.; Naumovski, A.L.; Langhoff, P.; Horvath, G.L.; Jin, T.; Xiao, T.S.; Garbi, N.; Agrawal, S.; Kolbeck, R.; Latz, E. RAGE Enhances TLR Responses through Binding and Internalization of RNA. J. Immunol. 2016, 197, 4118–4126. [Google Scholar] [CrossRef]
- Gao, J.X.; Issekutz, A.C. Mac-1 (CD11b/CD18) Is the Predominant Beta 2 (CD18) Integrin Mediating Human Neutrophil Migration through Synovial and Dermal Fibroblast Barriers. Immunology 1996, 88, 463–470. [Google Scholar] [CrossRef]
- Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. beta2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. [Google Scholar] [CrossRef]
- Varga, G.; Balkow, S.; Wild, M.K.; Stadtbaeumer, A.; Krummen, M.; Rothoeft, T.; Higuchi, T.; Beissert, S.; Wethmar, K.; Scharffetter-Kochanek, K.; et al. Active MAC-1 (CD11b/CD18) on DCs Inhibits Full T-cell Activation. Blood 2007, 109, 661–669. [Google Scholar] [CrossRef]
- Teh, B.K.; Yeo, J.G.; Chern, L.M.; Lu, J. C1q Regulation of Dendritic Cell Development from Monocytes with Distinct Cytokine Production and T Cell Stimulation. Mol. Immunol. 2011, 48, 1128–1138. [Google Scholar] [CrossRef]
- Paidassi, H.; Tacnet-Delorme, P.; Garlatti, V.; Darnault, C.; Ghebrehiwet, B.; Gaboriaud, C.; Arlaud, G.J.; Frachet, P. C1q binds Phosphatidylserine and Likely Acts as a Multiligand-Bridging Molecule in Apoptotic Cell Recognition. J. Immunol. 2008, 180, 2329–2338. [Google Scholar] [CrossRef]
- Friggeri, A.; Banerjee, S.; Biswas, S.; de Freitas, A.; Liu, G.; Bierhaus, A.; Abraham, E. Participation of the Receptor for Advanced Glycation End Products in Efferocytosis. J. Immunol. 2011, 186, 6191–6198. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Kubo, H.; Morimoto, K.; Fujino, N.; Suzuki, T.; Takahasi, T.; Yamada, M.; Yamaya, M.; Maekawa, T.; Yamamoto, Y.; et al. Receptor for Advanced Glycation End Products Binds to Phosphatidylserine and Assists in the Clearance of Apoptotic Cells. EMBO Rep. 2011, 12, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Friggeri, A.; Liu, G.; Abraham, E. The C-Terminal Acidic Tail Is Responsible for the Inhibitory Effects of HMGB1 on Efferocytosis. J. Leukoc. Biol. 2010, 88, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Op den Kamp, J.A. Lipid Asymmetry in Membranes. Annu. Rev. Biochem. 1979, 48, 47–71. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the Dogma–Phosphatidylserine in Non-Apoptotic Cell Death. Cell Commun. Signal. 2019, 17, 139. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE Mediates Amyloid-Beta Peptide Transport across the Blood-Brain Barrier and Accumulation in Brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Li, M.; Shang, D.S.; Zhao, W.D.; Tian, L.; Li, B.; Fang, W.G.; Zhu, L.; Man, S.M.; Chen, Y.H. Amyloid Beta Interaction with Receptor for Advanced Glycation End Products Up-Regulates Brain Endothelial CCR5 Expression and Promotes T Cells Crossing the Blood-Brain Barrier. J. Immunol. 2009, 182, 5778–5788. [Google Scholar] [CrossRef]
- Sousa, M.M.; Yan, S.D.; Stern, D.; Saraiva, M.J. Interaction of the Receptor for Advanced Glycation End Products (RAGE) with Transthyretin Triggers Nuclear Transcription Factor kB (NF-kB) Activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef]
- Monu; Agnihotri, P.; Saquib, M.; Sarkar, A.; Chakraborty, D.; Kumar, U.; Biswas, S. Transthyretin and Receptor for Advanced Glycation End Product’s Differential Levels Associated with the Pathogenesis of Rheumatoid Arthritis. J. Inflamm. Res. 2021, 14, 5581–5596. [Google Scholar] [CrossRef] [PubMed]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 Proteins in Mouse and Man: From Evolution to Function and Pathology (Including an Update of the Nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 Proteins in Health and Disease. Biochim. Et Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef]
- Sreejit, G.; Flynn, M.C.; Patil, M.; Krishnamurthy, P.; Murphy, A.J.; Nagareddy, P.R. S100 Family Proteins in Inflammation and Beyond. Adv. Clin. Chem. 2020, 98, 173–231. [Google Scholar] [CrossRef]
- Most, P.; Bernotat, J.; Ehlermann, P.; Pleger, S.T.; Reppel, M.; Borries, M.; Niroomand, F.; Pieske, B.; Janssen, P.M.; Eschenhagen, T.; et al. S100A1: A Regulator of Myocardial Contractility. Proc. Natl. Acad. Sci. USA 2001, 98, 13889–13894. [Google Scholar] [CrossRef]
- Rohde, D.; Schon, C.; Boerries, M.; Didrihsone, I.; Ritterhoff, J.; Kubatzky, K.F.; Volkers, M.; Herzog, N.; Mahler, M.; Tsoporis, J.N.; et al. S100A1 is Released from Ischemic Cardiomyocytes and Signals Myocardial Damage Via Toll-Like Receptor 4. EMBO Mol. Med. 2014, 6, 778–794. [Google Scholar] [CrossRef]
- Zhou, Y.; Zha, Y.; Yang, Y.; Ma, T.; Li, H.; Liang, J. S100 Proteins in Cardiovascular Diseases. Mol. Med. 2023, 29, 68. [Google Scholar] [CrossRef]
- Kizawa, K.; Tsuchimoto, S.; Hashimoto, K.; Uchiwa, H. Gene Expression of Mouse S100A3, a Cysteine-Rich Calcium-Binding Protein, in Developing Hair Follicle. J. Investig. Dermatol. 1998, 111, 879–886. [Google Scholar] [CrossRef]
- Kizawa, K.; Uchiwa, H.; Murakami, U. Highly-Expressed S100A3, a Calcium-Binding Protein, in Human Hair Cuticle. Biochim. Biophys. Acta 1996, 1312, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Kizawa, K.; Jinbo, Y.; Inoue, T.; Takahara, H.; Unno, M.; Heizmann, C.W.; Izumi, Y. Human S100A3 Tetramerization Propagates Ca(2+)/Zn(2+) Binding States. Biochim. Biophys. Acta 2013, 1833, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E. Measuring Binding of S100 Proteins to RAGE by Surface Plasmon Resonance. Methods Mol. Biol. 2013, 963, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 Proteins: Dual-Function Alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Sunderkotter, C.; Foell, D.; Vogl, T.; Roth, J. The Endogenous Toll-like Receptor 4 Agonist S100A8/S100A9 (calprotectin) as Innate Amplifier of Infection, Autoimmunity, and Cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef]
- Muoio, M.G.; Talia, M.; Lappano, R.; Sims, A.H.; Vella, V.; Cirillo, F.; Manzella, L.; Giuliano, M.; Maggiolini, M.; Belfiore, A.; et al. Activation of the S100A7/RAGE Pathway by IGF-1 Contributes to Angiogenesis in Breast Cancer. Cancers 2021, 13, 621. [Google Scholar] [CrossRef]
- Rojas, A.; Araya, P.; Romero, J.; Delgado-Lopez, F.; Gonzalez, I.; Anazco, C.; Perez-Castro, R. Skewed Signaling through the Receptor for Advanced Glycation End-Products Alters the Proinflammatory Profile of Tumor-Associated Macrophages. Cancer Microenviron. 2018, 11, 97–105. [Google Scholar] [CrossRef]
- Rojas, A.; Schneider, I.; Lindner, C.; Gonzalez, I.; Morales, M.A. The RAGE/Multiligand Axis: A New Actor in Tumor Biology. Biosci. Rep. 2022, 42, BSR20220395. [Google Scholar] [CrossRef]
- Srikrishna, G.; Freeze, H.H. Endogenous Damage-Associated Molecular Pattern Molecules at the Crossroads of Inflammation and Cancer. Neoplasia 2009, 11, 615–628. [Google Scholar] [CrossRef]
- Pan, S.C.; Li, C.Y.; Kuo, C.Y.; Kuo, Y.Z.; Fang, W.Y.; Huang, Y.H.; Hsieh, T.C.; Kao, H.Y.; Kuo, Y.; Kang, Y.R.; et al. The P53-S100A2 Positive Feedback Loop Negatively Regulates Epithelialization in Cutaneous Wound Healing. Sci. Rep. 2018, 8, 5458. [Google Scholar] [CrossRef]
- Yoshioka, M.; Sawada, Y.; Saito-Sasaki, N.; Yoshioka, H.; Hama, K.; Omoto, D.; Ohmori, S.; Okada, E.; Nakamura, M. High S100A2 Expression in Keratinocytes in Patients with Drug Eruption. Sci. Rep. 2021, 11, 5493. [Google Scholar] [CrossRef] [PubMed]
- Poachanukoon, O.; Roytrakul, S.; Koontongkaew, S. A Shotgun Proteomic Approach Reveals Novel Potential Salivary Protein Biomarkers for Asthma. J. Asthma 2022, 59, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Sugino, H.; Sawada, Y. Influence of S100A2 in Human Diseases. Diagnostics 2022, 12, 1756. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Mirmohammadsadegh, A.; Walz, M.; Lysa, B.; Tartler, U.; Remus, R.; Hengge, U.; Michel, G.; Ruzicka, T. Molecular Cloning and Characterization of Alternatively Spliced mRNA Isoforms from Psoriatic Skin Encoding a Novel Member of the S100 Family. FASEB J. 2003, 17, 1969–1971. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 Proteins in Cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef]
- Li, Z.H.; Bresnick, A.R. The S100A4 Metastasis Factor Regulates Cellular Motility Via a Direct Interaction with Myosin-IIA. Cancer Res. 2006, 66, 5173–5180. [Google Scholar] [CrossRef]
- Boye, K.; Maelandsmo, G.M. S100A4 and Metastasis: A Small Actor Playing Many Roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef]
- Ambartsumian, N.; Klingelhofer, J.; Grigorian, M. The Multifaceted S100A4 Protein in Cancer and Inflammation. Methods Mol. Biol. 2019, 1929, 339–365. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Liu, S.; Qin, Z. Extracellular S100A4 as a Key Player in Fibrotic Diseases. J. Cell Mol. Med. 2020, 24, 5973–5983. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Milani, M.; Apolloni, S. S100A4 in the Physiology and Pathology of the Central and Peripheral Nervous System. Cells 2021, 10, 798. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, S.; Gu, J.; Tian, T.; Yuan, Q.; Jia, J.; Qin, Z.; Chen, Z. S100A4 Promotes Colon Inflammation and Colitis-Associated Colon Tumorigenesis. Oncoimmunology 2018, 7, e1461301. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ma, J.; Zhu, D.; Wang, Z.; Li, Y.; He, X.; Zhang, G.; Kang, X. The Role of S100A6 in Human Diseases: Molecular Mechanisms and Therapeutic Potential. Biomolecules 2023, 13, 1139. [Google Scholar] [CrossRef] [PubMed]
- Edgeworth, J.; Gorman, M.; Bennett, R.; Freemont, P.; Hogg, N. Identification of p8,14 as a Highly Abundant Heterodimeric Calcium Binding Protein Complex of Myeloid Cells. J. Biol. Chem. 1991, 266, 7706–7713. [Google Scholar] [CrossRef]
- Dale, I.; Fagerhol, M.K.; Naesgaard, I. Purification and Partial Characterization of a Highly Immunogenic Human Leukocyte Protein, the L1 Antigen. Eur. J. Biochem. 1983, 134, 850–858. [Google Scholar] [CrossRef]
- Curran, C.S.; Bertics, P.J. Human Eosinophils Express RAGE, Produce RAGE Ligands, Exhibit PKC-Delta Phosphorylation and Enhanced Viability in Response to the RAGE Ligand, S100B. Int. Immunol. 2011, 23, 713–728. [Google Scholar] [CrossRef]
- Gebhardt, C.; Nemeth, J.; Angel, P.; Hess, J. S100A8 and S100A9 in Inflammation and Cancer. Biochem. Pharmacol. 2006, 72, 1622–1631. [Google Scholar] [CrossRef]
- Basso, D.; Fogar, P.; Plebani, M. The S100A8/A9 complex reduces CTLA4 Expression by Immature Myeloid Cells: Implications for Pancreatic Cancer-Driven Immunosuppression. Oncoimmunology 2013, 2, e24441. [Google Scholar] [CrossRef]
- Ghavami, S.; Chitayat, S.; Hashemi, M.; Eshraghi, M.; Chazin, W.J.; Halayko, A.J.; Kerkhoff, C. S100A8/A9: A Janus-Faced Molecule in Cancer Therapy and Tumorgenesis. Eur. J. Pharmacol. 2009, 625, 73–83. [Google Scholar] [CrossRef]
- Miyamoto, S.; Ueda, M.; Ikemoto, M.; Naruko, T.; Itoh, A.; Tamaki, S.; Nohara, R.; Terasaki, F.; Sasayama, S.; Fujita, M. Increased Serum Levels and Expression of S100A8/A9 Complex in Infiltrated Neutrophils in Atherosclerotic Plaque of Unstable Angina. Heart 2008, 94, 1002–1007. [Google Scholar] [CrossRef]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef]
- Soyfoo, M.S.; Roth, J.; Vogl, T.; Pochet, R.; Decaux, G. Phagocyte-Specific S100A8/A9 Protein levels During Disease Exacerbations and Infections in Systemic Lupus Erythematosus. J. Rheumatol. 2009, 36, 2190–2194. [Google Scholar] [CrossRef] [PubMed]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Yamamoto, H.; Chazin, W.J.; Nakatani, Y.; Yui, S.; Makino, H. The S100A8/A9 Heterodimer Amplifies Proinflammatory Cytokine Production by Macrophages Via Activation of Nuclear Factor Kappa B and P38 Mitogen-Activated Protein Kinase in Rheumatoid Arthritis. Arthritis Res. Ther. 2006, 8, R69. [Google Scholar] [CrossRef] [PubMed]
- van Lent, P.L.; Grevers, L.; Blom, A.B.; Sloetjes, A.; Mort, J.S.; Vogl, T.; Nacken, W.; van den Berg, W.B.; Roth, J. Myeloid Related Proteins S100A8/S100A9 Regulate Joint Inflammation and Cartilage Destruction During Antigen-Induced Arthritis. Ann. Rheum. Dis. 2007, 67, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100A8/A9: From Basic Science to Clinical Application. Pharmacol. Ther. 2016, 167, 120–131. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef]
- Kinoshita, R.; Sato, H.; Yamauchi, A.; Takahashi, Y.; Inoue, Y.; Sumardika, I.W.; Chen, Y.; Tomonobu, N.; Araki, K.; Shien, K.; et al. ExSSSRs (Extracellular S100 Soil Sensor Receptors)-Fc Fusion Proteins Work as Prominent Decoys to S100A8/A9-Induced Lung Tropic Cancer Metastasis. Int. J. Cancer 2019, 144, 3138–3145. [Google Scholar] [CrossRef]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at Low Concentration Promotes Tumor Cell Growth Via RAGE Ligation and MAP Kinase-Dependent Pathway. J. Leukoc. Biol. 2008, 83, 1484–1492. [Google Scholar] [CrossRef]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, Carboxylated Glycans and S100A8/A9 Play Essential Roles in Colitis-Associated Carcinogenesis. Carcinogenesis 2008, 29, 2035–2043. [Google Scholar] [CrossRef]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. MRP8 and MRP14 Are Endogenous Activators of Toll-Like Receptor 4, Promoting Lethal, Endotoxin-Induced Shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, C.; Shao, R.; Xu, Y.; Zhao, W. The Functions and Regulatory Pathways of S100A8/A9 and Its Receptors in Cancers. Front. Pharmacol. 2023, 14, 1187741. [Google Scholar] [CrossRef]
- Miles, L.A.; Parmer, R.J. S100A10: A Complex Inflammatory Role. Blood 2010, 116, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, P.A.; Surette, A.P.; Liwski, R.S.; Svenningsson, P.; Waisman, D.M. S100A10 Regulates Plasminogen-Dependent Macrophage Invasion. Blood 2010, 116, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Propper, C.; Hartmann, M.; Strey, A.; Strupat, K.; van den Bos, C.; Sorg, C.; Roth, J. S100A12 is Expressed Exclusively by Granulocytes and Acts Independently from MRP8 and MRP14. J. Biol. Chem. 1999, 274, 25291–25296. [Google Scholar] [CrossRef] [PubMed]
- Meijer, B.; Gearry, R.B.; Day, A.S. The Role of S100A12 as a Systemic Marker of Inflammation. Int. J. Inflam. 2012, 2012, 907078. [Google Scholar] [CrossRef]
- Carvalho, A.; Lu, J.; Francis, J.D.; Moore, R.E.; Haley, K.P.; Doster, R.S.; Townsend, S.D.; Johnson, J.G.; Damo, S.M.; Gaddy, J.A. S100A12 in Digestive Diseases and Health: A Scoping Review. Gastroenterol. Res. Pract. 2020, 2020, 2868373. [Google Scholar] [CrossRef]
- Michetti, F.; Clementi, M.E.; Di Liddo, R.; Valeriani, F.; Ria, F.; Rende, M.; Di Sante, G.; Romano Spica, V. The S100B Protein: A Multifaceted Pathogenic Factor More Than a Biomarker. Int. J. Mol. Sci. 2023, 24, 9605. [Google Scholar] [CrossRef]
- Van Eldik, L.J.; Wainwright, M.S. The Janus Face of Glial-Derived S100B: Beneficial and Detrimental Functions in the Brain. Restor. Neurol. Neurosci. 2003, 21, 97–108. [Google Scholar]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s Double Life: Intracellular Regulator and Extracellular Signal. Biochim. Biophys. Acta 2009, 1793, 1008–1022. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Arendash, G.W.; Horikoshi-Sakuraba, Y.; Tan, J.; Town, T. Overexpression of Human S100B Exacerbates Cerebral Amyloidosis and Gliosis in the Tg2576 Mouse Model of Alzheimer’s Disease. Glia 2010, 58, 300–314. [Google Scholar] [CrossRef]
- Roltsch, E.; Holcomb, L.; Young, K.A.; Marks, A.; Zimmer, D.B. PSAPP Mice Exhibit Regionally Selective Reductions in Gliosis and Plaque Deposition in Response to S100B Ablation. J. Neuroinflamm. 2010, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Barros, C.; Barateiro, A.; Neto, A.; Soromenho, B.; Basto, A.P.; Mateus, J.M.; Xapelli, S.; Sebastiao, A.M.; Brites, D.; Graca, L.; et al. S100B Inhibition Protects from Chronic Experimental Autoimmune Encephalomyelitis. Brain Commun. 2022, 4, fcac076. [Google Scholar] [CrossRef]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Turco, F.; Steardo, L.; Cuomo, R. S100B Protein in the Gut: The Evidence for Enteroglial-Sustained Intestinal Inflammation. World J. Gastroenterol. 2011, 17, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, J.S.; Morris, V.K.; Cardoso, I.; Leal, S.S.; Martinez, J.; Botelho, H.M.; Gobl, C.; David, R.; Kierdorf, K.; Alemi, M.; et al. The Neuronal S100B Protein is a Calcium-Tuned Suppressor of Amyloid-Beta Aggregation. Sci. Adv. 2018, 4, eaaq1702. [Google Scholar] [CrossRef] [PubMed]
- Moreira, G.G.; Cantrelle, F.X.; Quezada, A.; Carvalho, F.S.; Cristovao, J.S.; Sengupta, U.; Puangmalai, N.; Carapeto, A.P.; Rodrigues, M.S.; Cardoso, I.; et al. Dynamic Interactions and Ca(2+)-Binding Modulate the Holdase-Type Chaperone Activity of S100B Preventing Tau Aggregation and Seeding. Nat. Commun. 2021, 12, 6292. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Manfredi, A.A.; Capobianco, A.; Esposito, A.; De Cobelli, F.; Canu, T.; Monno, A.; Raucci, A.; Sanvito, F.; Doglioni, C.; Nawroth, P.P.; et al. Maturing Dendritic Cells Depend on RAGE for in Vivo Homing to Lymph Nodes. J. Immunol. 2008, 180, 2270–2275. [Google Scholar] [CrossRef]
- Rovere-Querini, P.; Capobianco, A.; Scaffidi, P.; Valentinis, B.; Catalanotti, F.; Giazzon, M.; Dumitriu, I.E.; Muller, S.; Iannacone, M.; Traversari, C.; et al. HMGB1 is an Endogenous Immune Adjuvant Released by Necrotic Cells. EMBO Rep. 2004, 5, 825–830. [Google Scholar] [CrossRef]
- Messmer, D.; Yang, H.; Telusma, G.; Knoll, F.; Li, J.; Messmer, B.; Tracey, K.J.; Chiorazzi, N. High Mobility Group Box Protein 1: An Endogenous Signal for Dendritic Cell Maturation and Th1 Polarization. J. Immunol. 2004, 173, 307–313. [Google Scholar] [CrossRef]
- Mu, L.; Zhang, Y.; Sun, B.; Wang, J.; Xie, X.; Li, N.; Zhang, J.; Kong, Q.; Liu, Y.; Han, Z.; et al. Activation of the Receptor for Advanced Glycation End Products (RAGE) Exacerbates Experimental Autoimmune Myasthenia Gravis Symptoms. Clin. Immunol. 2011, 141, 36–48. [Google Scholar] [CrossRef]
- Akirav, E.M.; Preston-Hurlburt, P.; Garyu, J.; Henegariu, O.; Clynes, R.; Schmidt, A.M.; Herold, K.C. RAGE Expression in Human T cells: A Link Between Environmental Factors and Adaptive Immune Responses. PLoS ONE 2012, 7, e34698. [Google Scholar] [CrossRef] [PubMed]
- Durning, S.P.; Preston-Hurlburt, P.; Clark, P.R.; Xu, D.; Herold, K.C.; Grp, T.D.T.S. The Receptor for Advanced Glycation Endproducts Drives T Cell Survival and Inflammation in Type 1 Diabetes Mellitus. J. Immunol. 2016, 197, 3076–3085. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Preston-Hurlburt, P.; Philbrick, W.; Betancur, G.; Korah, M.; Lucas, C.; Herold, K.C. The Receptor for Advanced Glycation Endproducts (RAGE) Modulates T Cell Signaling. PLoS ONE 2020, 15, e0236921. [Google Scholar] [CrossRef] [PubMed]
- Strohbuecker, L.; Koenen, H.; van Rijssen, E.; van Cranenbroek, B.; Fasse, E.; Joosten, I.; Korber, A.; Bergmann, C. Increased Dermal Expression of Chromatin-Associated Protein HMGB1 and Concomitant T-cell Expression of the DNA RAGE in Patients with Psoriasis Vulgaris. Psoriasis 2019, 9, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Austermann, J.; Roth, J.; Barczyk-Kahlert, K. The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation. Cells 2022, 11, 1979. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in Immunoregulation and Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Mosser, D.M.; Hamidzadeh, K.; Goncalves, R. Macrophages and the Maintenance of Homeostasis. Cell. Mol. Immunol. 2021, 18, 579–587. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: Their Untold Story in T Cell Activation and Function. Int. Rev. Cell Mol. Biol. 2019, 342, 73–93. [Google Scholar] [CrossRef]
- Su, C.; Jia, S.; Ma, Z.; Zhang, H.; Wei, L.; Liu, H. HMGB1 Promotes Lymphangiogenesis through the Activation of RAGE on M2 Macrophages in Laryngeal Squamous Cell Carcinoma. Dis. Markers 2022, 2022, 4487435. [Google Scholar] [CrossRef]
- He, J.; Wei, L.; Tan, S.; Liang, B.; Liu, J.; Lu, L.; Wang, T.; Wang, J.; Huang, Y.; Chen, Z.; et al. Macrophage RAGE Deficiency Prevents Myocardial Fibrosis by Repressing Autophagy-Mediated Macrophage Alternative Activation. FASEB J. 2023, 37, e23259. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage Activation and Polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Brett, J.; Mora, R.; Nowygrod, R.; Stern, D. Regulation of Human Mononuclear Phagocyte Migration by Cell Surface-Binding Proteins for Advanced Glycation End Products. J. Clin. Investig. 1993, 91, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M. 22016 ATVB Plenary Lecture: Receptor for Advanced Glycation Endproducts and Implications for the Pathogenesis and Treatment of Cardiometabolic Disorders: Spotlight on the Macrophage. Arter. Thromb. Vasc. Biol. 2017, 37, 613–621. [Google Scholar] [CrossRef]
- Osonoi, S.; Mizukami, H.; Takeuchi, Y.; Sugawa, H.; Ogasawara, S.; Takaku, S.; Sasaki, T.; Kudoh, K.; Ito, K.; Sango, K.; et al. RAGE Activation in Macrophages and Development of Experimental Diabetic Polyneuropathy. JCI Insight 2022, 7, e160555. [Google Scholar] [CrossRef]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-kappaB Pathway. BioMed. Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef]
- Juranek, J.K.; Geddis, M.S.; Song, F.; Zhang, J.; Garcia, J.; Rosario, R.; Yan, S.F.; Brannagan, T.H.; Schmidt, A.M. RAGE Deficiency Improves Postinjury Sciatic Nerve Regeneration in Type 1 Diabetic Mice. Diabetes 2013, 62, 931–943. [Google Scholar] [CrossRef]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE Axis in Neuroinflammation, Neurodegeneration and Its Emerging Role in the Pathogenesis of Amyotrophic Lateral Sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Harashima, A.; Saito, H.; Tsuneyama, K.; Munesue, S.; Motoyoshi, S.; Han, D.; Watanabe, T.; Asano, M.; Takasawa, S.; et al. Septic Shock is Associated with Receptor for Advanced Glycation end Products Ligation of LPS. J. Immunol. 2011, 186, 3248–3257. [Google Scholar] [CrossRef]
- Daffu, G.; Shen, X.; Senatus, L.; Thiagarajan, D.; Abedini, A.; Hurtado Del Pozo, C.; Rosario, R.; Song, F.; Friedman, R.A.; Ramasamy, R.; et al. RAGE Suppresses ABCG1-Mediated Macrophage Cholesterol Efflux in Diabetes. Diabetes 2015, 64, 4046–4060. [Google Scholar] [CrossRef]
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. HMGB1 Develops Enhanced Proinflammatory Activity by Binding to Cytokines. J. Immunol. 2008, 180, 2531–2537. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Du, Z.; Shu, X.; Zhu, L.; Wu, J.; Gao, Q.; Wang, L.; Chen, N.; Li, Y.; Luo, M.; et al. Role of RAGE in Obesity-Induced Adipose Tissue Inflammation and Insulin Resistance. Cell Death Discov. 2021, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. RAGE Potentiates Abeta-Induced Perturbation of Neuronal Function in Transgenic Mice. Embo J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Dalpke, A.; Mracsko, E.; Antoine, D.J.; Roth, S.; Zhou, W.; Yang, H.; Na, S.Y.; Akhisaroglu, M.; Fleming, T.; et al. DAMP Signaling Is a Key Pathway Inducing Immune Modulation After Brain Injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 Receptor RAGE Mediates Ischemic Brain Damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef]
- Gajewska-Naryniecka, A.; Szwedowicz, U.; Lapinska, Z.; Rudno-Rudzinska, J.; Kielan, W.; Kulbacka, J. Irreversible Electroporation in Pancreatic Cancer-An Evolving Experimental and Clinical Method. Int. J. Mol. Sci. 2023, 24, 4381. [Google Scholar] [CrossRef]
- Zhao, J.; Wen, X.; Tian, L.; Li, T.; Xu, C.; Wen, X.; Melancon, M.P.; Gupta, S.; Shen, B.; Peng, W.; et al. Irreversible Electroporation Reverses Resistance to Immune Checkpoint Blockade in Pancreatic Cancer. Nat. Commun. 2019, 10, 899. [Google Scholar] [CrossRef]
- Lutterloh, E.C.; Opal, S.M.; Pittman, D.D.; Keith, J.C., Jr.; Tan, X.Y.; Clancy, B.M.; Palmer, H.; Milarski, K.; Sun, Y.; Palardy, J.E.; et al. Inhibition of the RAGE Products Increases Survival in Experimental Models of Severe Sepsis and Systemic Infection. Crit. Care 2007, 11, R122. [Google Scholar] [CrossRef]
- Prantner, D.; Nallar, S.; Richard, K.; Spiegel, D.; Collins, K.D.; Vogel, S.N. Classically Activated Mouse Macrophages Produce Methylglyoxal that Induces a TLR4- and RAGE-Independent Proinflammatory Response. J. Leukoc. Biol. 2021, 109, 605–619. [Google Scholar] [CrossRef]
- Lopez-Diez, R.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Cellular Mechanisms and Consequences of Glycation in Atherosclerosis and Obesity. Biochim. Biophys. Acta 2016, 1862, 2244–2252. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Cipollone, F.; Iezzi, A.; Fazia, M.; Zucchelli, M.; Pini, B.; Cuccurullo, C.; De Cesare, D.; De Blasis, G.; Muraro, R.; Bei, R.; et al. The Receptor RAGE as a Progression Factor Amplifying Arachidonate-Dependent Inflammatory and Proteolytic Response in Human Atherosclerotic Plaques: Role of Glycemic Control. Circulation 2003, 108, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Kolodgie, F.D.; Zieske, A.; Fowler, D.R.; Weber, D.K.; Varghese, P.J.; Farb, A.; Virmani, R. Morphologic Findings of Coronary Atherosclerotic Plaques in Diabetics: A Postmortem Study. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ishida, T.; Yasuda, T.; Kojima, Y.; Honjo, T.; Yamamoto, Y.; Yamamoto, H.; Ishibashi, S.; Hirata, K.; Hayashi, Y. RAGE Mediates Oxidized LDL-Induced Pro-Inflammatory Effects and Atherosclerosis in Non-Diabetic LDL Receptor-Deficient Mice. Cardiovasc. Res. 2009, 82, 371–381. [Google Scholar] [CrossRef]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and Inflammatory Stresses Mediate Atherosclerosis Via RAGE and Its Ligands in Apoe-/-Mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C.; et al. RAGE Blockade Stabilizes Established Atherosclerosis in Diabetic Apolipoprotein E-null Mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef]
- Shirasawa, M.; Fujiwara, N.; Hirabayashi, S.; Ohno, H.; Iida, J.; Makita, K.; Hata, Y. Receptor for Advanced Glycation End-Products Is a Marker of Type I Lung Alveolar Cells. Genes. Cells 2004, 9, 165–174. [Google Scholar] [CrossRef]
- Dahlin, K.; Mager, E.M.; Allen, L.; Tigue, Z.; Goodglick, L.; Wadehra, M.; Dobbs, L. Identification of Genes Differentially Expressed in Rat Alveolar Type I Cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 309–316. [Google Scholar] [CrossRef]
- Englert, J.M.; Hanford, L.E.; Kaminski, N.; Tobolewski, J.M.; Tan, R.J.; Fattman, C.L.; Ramsgaard, L.; Richards, T.J.; Loutaev, I.; Nawroth, P.P.; et al. A Role for the Receptor for Advanced Glycation End Products in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2008, 172, 583–591. [Google Scholar] [CrossRef]
- Ramsgaard, L.; Englert, J.M.; Tobolewski, J.; Tomai, L.; Fattman, C.L.; Leme, A.S.; Kaynar, A.M.; Shapiro, S.D.; Enghild, J.J.; Oury, T.D. The Role of the Receptor for Advanced Glycation End-Products in a Murine Model of Silicosis. PLoS ONE 2010, 5, e9604. [Google Scholar] [CrossRef]
- Englert, J.M.; Kliment, C.R.; Ramsgaard, L.; Milutinovic, P.S.; Crum, L.; Tobolewski, J.M.; Oury, T.D. Paradoxical Function for the Receptor for Advanced Glycation End Products in Mouse Models of Pulmonary Fibrosis. Int. J. Clin. Exp. Pathol. 2011, 4, 241–254. [Google Scholar] [PubMed]
- He, M.; Kubo, H.; Ishizawa, K.; Hegab, A.E.; Yamamoto, Y.; Yamamoto, H.; Yamaya, M. The Role of the Receptor for Advanced Glycation End-Products in Lung Fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1427–L1436. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Penza, F.; Bianchi, N.; Olivieri, C.; Bennett, D.; Prasse, A.; Rottoli, P. Controversial Role of RAGE in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Respir. Physiol. Neurobiol. 2009, 165, 119–120, author reply 121–112. [Google Scholar] [CrossRef] [PubMed]
- Queisser, M.A.; Kouri, F.M.; Konigshoff, M.; Wygrecka, M.; Schubert, U.; Eickelberg, O.; Preissner, K.T. Loss of RAGE in Pulmonary Fibrosis: Molecular Relations to Functional Changes in Pulmonary Cell Types. Am. J. Respir. Cell Mol. Biol. 2008, 39, 337–345. [Google Scholar] [CrossRef]
- Radtke, D.; Voehringer, D. Granulocyte Development, Tissue Recruitment, and Function During Allergic Inflammation. Eur. J. Immunol. 2023, 53, e2249977. [Google Scholar] [CrossRef]
- Watanabe, H.; Kubo, M.; Taniguchi, A.; Asano, Y.; Hiramatsu-Asano, S.; Ohashi, K.; Zeggar, S.; Katsuyama, E.; Katsuyama, T.; Sunahori-Watanabe, K.; et al. Amelioration of Nephritis in Receptor for Advanced Glycation End-Products (RAGE)-Deficient Lupus-Prone Mice through Neutrophil Extracellular Traps. Clin. Immunol. 2023, 250, 109317. [Google Scholar] [CrossRef]
- Collison, K.S.; Parhar, R.S.; Saleh, S.S.; Meyer, B.F.; Kwaasi, A.A.; Hammami, M.M.; Schmidt, A.M.; Stern, D.M.; Al-Mohanna, F.A. RAGE-Mediated Neutrophil Dysfunction Is Evoked by Advanced Glycation End Products (AGEs). J. Leukoc. Biol. 2002, 71, 433–444. [Google Scholar] [CrossRef]
- Toure, F.; Zahm, J.M.; Garnotel, R.; Lambert, E.; Bonnet, N.; Schmidt, A.M.; Vitry, F.; Chanard, J.; Gillery, P.; Rieu, P. Receptor for Advanced Glycation End-Products (RAGE) Modulates Neutrophil Adhesion and Migration on Glycoxidated Extracellular matrix. Biochem. J. 2008, 416, 255–261. [Google Scholar] [CrossRef]
- Zen, K.; Chen, C.X.; Chen, Y.T.; Wilton, R.; Liu, Y. Receptor for Advanced Glycation Endproducts Mediates Neutrophil Migration Across Intestinal Epithelium. J. Immunol. 2007, 178, 2483–2490. [Google Scholar] [CrossRef]
- Tatsiy, O.; de Carvalho Oliveira, V.; Mosha, H.T.; McDonald, P.P. Early and Late Processes Driving NET Formation, and the Autocrine/Paracrine Role of Endogenous RAGE Ligands. Front. Immunol. 2021, 12, 675315. [Google Scholar] [CrossRef]
- Allam, V.; Faiz, A.; Lam, M.; Rathnayake, S.N.H.; Ditz, B.; Pouwels, S.D.; Brandsma, C.A.; Timens, W.; Hiemstra, P.S.; Tew, G.W.; et al. RAGE and TLR4 differentially regulate airway hyperresponsiveness: Implications for Copd. Allergy 2021, 76, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, K.; Huang, B.; Li, R.; Wang, X.; Zhang, H.; Tang, H.; Chen, X. The Receptor for Advanced Glycation End Products Mediates Dysfunction of Airway Epithelial Barrier in a Lipopolysaccharides-Induced Murine Acute Lung Injury Model. Int. Immunopharmacol. 2021, 93, 107419. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, R.; Herzog, G.I.; DeMarco, R.A.; Beer-Stolz, D.; Lee, J.J.; Rubartelli, A.; Schrezenmeier, H.; Lotze, M.T. Eosinophils Oxidize Damage-Associated Molecular Pattern Molecules Derived from Stressed Cells. J. Immunol. 2009, 183, 5023–5031. [Google Scholar] [CrossRef] [PubMed]
- Perkins, T.N.; Oczypok, E.A.; Milutinovic, P.S.; Dutz, R.E.; Oury, T.D. RAGE-Dependent VCAM-1 Expression in the Lung Endothelium Mediates IL-33-Induced Allergic Airway Inflammation. Allergy 2019, 74, 89–99. [Google Scholar] [CrossRef]
- Perkins, T.N.; Oczypok, E.A.; Dutz, R.E.; Donnell, M.L.; Myerburg, M.M.; Oury, T.D. The Receptor for Advanced Glycation End Products is a Critical Mediator of Type 2 Cytokine Signaling in the Lungs. J. Allergy Clin. Immunol. 2019, 144, 796–808.e712. [Google Scholar] [CrossRef]
- Milutinovic, P.S.; Alcorn, J.F.; Englert, J.M.; Crum, L.T.; Oury, T.D. The Receptor for Advanced Glycation End Products Is a central Mediator of Asthma Pathogenesis. Am. J. Pathol. 2012, 181, 1215–1225. [Google Scholar] [CrossRef]
- Oczypok, E.A.; Milutinovic, P.S.; Alcorn, J.F.; Khare, A.; Crum, L.T.; Manni, M.L.; Epperly, M.W.; Pawluk, A.M.; Ray, A.; Oury, T.D. Pulmonary Receptor for Advanced Glycation End-Products Promotes Asthma Pathogenesis through IL-33 and Accumulation of Group 2 Innate Lymphoid Cells. J. Allergy Clin. Immunol. 2015, 136, 747–756.e744. [Google Scholar] [CrossRef]
- Taniguchi, A.; Miyahara, N.; Waseda, K.; Kurimoto, E.; Fujii, U.; Tanimoto, Y.; Kataoka, M.; Yamamoto, Y.; Gelfand, E.W.; Yamamoto, H.; et al. Contrasting Roles for the Receptor for Advanced Glycation End-Products on Structural Cells in Allergic Airway Inflammation Vs. Airway Hyperresponsiveness. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L789–L800. [Google Scholar] [CrossRef]
- Ullah, M.A.; Loh, Z.; Gan, W.J.; Zhang, V.; Yang, H.; Li, J.H.; Yamamoto, Y.; Schmidt, A.M.; Armour, C.L.; Hughes, J.M.; et al. Receptor for Advanced Glycation End Products and Its Ligand High-Mobility Group Box-1 Mediate Allergic Airway Sensitization and Airway Inflammation. J. Allergy Clin. Immunol. 2014, 134, 440–450. [Google Scholar] [CrossRef]
- Manni, M.L.; Alcorn, J.F. Calprotectin-g the Lung during Type 2 Allergic Airway Inflammation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 405–407. [Google Scholar] [CrossRef]
- Han, K.; Suzukawa, M.; Yamaguchi, M.; Sugimoto, N.; Nakase, Y.; Toda, T.; Nagase, H.; Ohta, K. The in Vitro Effects of Advanced Glycation End Products on Basophil Functions. Int. Arch. Allergy Immunol. 2011, 155, 64–70. [Google Scholar] [CrossRef]
- Yan, L.; Li, Y.; Tan, T.; Qi, J.; Fang, J.; Guo, H.; Ren, Z.; Gou, L.; Geng, Y.; Cui, H.; et al. RAGE-TLR4 Crosstalk Is the Key Mechanism by Which High Glucose Enhances the Lipopolysaccharide-Induced Inflammatory Response in Primary Bovine Alveolar Macrophages. Int. J. Mol. Sci. 2023, 24, 7007. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Cell/Animal Model Used | Effect on Cells | Disease/Disease Model | References |
|---|---|---|---|---|
| AGE-β2-microglobulin | Human mononuclear phagocytes | Stimulates RAGE-dependent production of tumor necrosis factor alpha (TNF-α), oxidative stress, and chemotaxis | Dialysis-related amyloidosis | [61] |
| AGE-ovalbumin | Human immature dendritic cells (DC) | Enhances RAGE expression and NF-kB translocation to the nucleus Enhances IL-6 production | Food allergy | [62] |
| AGE-BSA | Monocyte-derived DCs | Enhances RAGE expression Promotes RAGE- and c-Jun N-terminal kinase (JNK)-dependent DC maturation Enhances the ability of DCs to activate T cells | Atherosclerosis | [63] |
| AGE-BSA | Rat peritoneal mast cells | Triggers RAGE-dependent exocytosis, histamine release and reactive oxygen species (ROS) production | Chronic inflammatory diseases | [64] |
| HMGB1/lipopolysaccharide (LPS) | Murine peritoneal macrophages | Triggers TNF-α and interleukin -6 (IL-6) secretion RAGE-dependent activation of p38 and NF-kB | Inflammation | [65] |
| HMGB1/nucleic acid | Murine DCs | Stimulates RAGE- and Toll like receptor -9 (TLR-9)-dependent cytokine production | Lupus | [66] |
| HMGB1 | Rat brain cortical neuron | Stimulates RAGE-dependent neurite outgrowth | Neuronal function | [67] |
| HMGB1 | Murine macrophage | Triggers RAGE-dependent macrophage and monocyte pyroptosis | Endotoxemia | [68] |
| HMGB1/LPS | Human monocyte | Enhances TNF-α production | Gram–bacteria induced sepsis | [69] |
| HMGB1/LPS | Murine peritoneal macrophages | RAGE-mediated HMGB1/LPS internalization HMGB1 enables LPS to activate intracellular caspase 11 and triggers pyroptosis. | Sepsis | [70] |
| Nucleic acid/HMGB1 | Human monocytic cell lines | Stimulates both RAGE-dependent activation and inhibition of inflammasome | Inflammation | [71] |
| Nucleic acid/HMGB1 | Murine RAW264.7 macrophages | Enhances RAGE-dependent activation of AKT, TNF-α release and cell death | Inflammation | [72] |
| C1q | U937-derived phagocytes | Triggers RAGE-dependent phagocytosis | Innate and adaptive immune response | [14] |
| C1q/HMGB1 | Peripheral blood monocytes | C1q inhibits HMGB1-induced monocyte activation C1q inhibits RAGE-dependent HMGB1 internalization C1q bridges RAGE and LAIR Promotes resolution of inflammation and the expression of resolvin D1 and D2 and lipoxin A4 | [73,74] | |
| MAC-1 | Mouse model | Stimulates RAGE-dependent leukocyte recruitment RAGE/MAC-1 interaction enhanced in the presence of S100B | Mouse model of acute peritonitis | [75] |
| MAC-1/HMGB1 | Mouse model | HMGB1 promotes RAGE-/MAC-1-dependent neutrophil recruitment HMGB1 enhances RAGE/MAC-1 interaction HMGB1 triggers RAGE- and MAC-1-dependent NF-κB activation | [76] | |
| Amyloid β (Aβ) | Microglia | Stimulates RAGE-dependent expression of M-CSF | Alzheimer’s Disease | [77] |
| Amyloid β (Aβ) | Neurons | Triggers RAGE- and NF-κB-dependent expression of M-CSF | Alzheimer’s Disease | [12] |
| Amyloid β (Aβ) | Brain tissue | Stimulates IL-1β and TNF-α production Enhances infiltration of microglia and astrocytes and Aβ accumulation Accelerates deterioration of spatial learning/memory abilities | Alzheimer’s Disease | [78] |
| Amyloid β (Aβ) | Human endothelial cells, neuronal cells, and microglia | Enhances RAGE-dependent oxidant stress and NF-κB activation in endothelial cells and neurons Stimulates RAGE-dependent migration of microglia and TNF-α expression | Alzheimer’s disease | [79] |
| S100A4 | Mouse model | Mediates macrophage recruitment and chemotaxis | Inflammation | [80] |
| S100A4 | Human peripheral blood samples from patients with rheumatoid arthritis | Stimulates TNF-α, IL-1β, and IL-6 secretion | Rheumatoid Arthritis | [81] |
| S100A6 | THP-1 monocytes | The TgSAG1 protein from T. gondii promotes the expression of TNF-α in a S100A6/vimentin- and PKC/NF-κB-dependent manner | Toxoplasma gondii infection | [82] |
| S100A6 | Mouse model of liver fibrosis | S100A6 triggers RAGE-dependent ERK phosphorylation and accelerates liver fibrosis | Liver fibrosis | [83] |
| S100A8, A9 | In vitro | Promotes leukocyte recruitment S100A9 induces MAC-1 expression | Inflammation | [84] |
| S100A8/A9 | Murine fibroblasts | Stimulates RAGE-dependent fibroblast proliferation and differentiation Increases collagen production Promotes RAGE-dependent NF-κB activity | Lung fibrosis | [85] |
| S100A8/A9 | Human macrophages | Stimulates TNFα, IL-1β, and IL-6 production | Inflammation | [86] |
| S100A8/A9 | Murine endothelial cells | Stimulates cell death (PANapoptosis) of endothelial cells | Mouse model of sepsis | [87] |
| S100A8/A9 | Cardiac fibroblasts | Triggers RAGE-dependent NF-kB activation Stimulates monocytes and cardiac fibroblasts migration | Inflammation-induced cardiac injury | [55] |
| S100A12 | Human monocytes | Stimulates TNF-α and IL-1β production Increases adhesion receptor expression in endothelial cells | Inflammation | [4] |
| S100A12 | Cord-blood-derived mast cells | Stimulates degranulation of mast cells Enhances RAGE-dependent TNF-α, IL-6, IL-8, MCP-1, and MIP-1β secretion | Inflammation | [88] |
| S100A12 | Human bronchial epithelial cells | Stimulates RAGE-dependent secretion of the MUC5AC mucin | Lung inflammation | [56]. |
| S100B | neurons | Neurotrophic effect at low concentration Neurotoxic effect at high concentration | Neuroinflammation | [89] |
| S100B | Rat astrocyte | Stimulates nitric oxide (NO) production | Neuroinflammation | [90] |
| S100B | RAGE-transfected N18 neuroblastoma cells on HMGB1-coated plates | Stimulates RAGE- and NF-κB-dependent neurite outgrowth at low concentration but triggers cell apotosis at high concentration | Neuroinflammation | [91] |
| S100B | Neuronal stem cells | Stimulates RAGE-dependent tau hyperphosphorylation through increases in JNK, AP-1/c-Jun, Dickopff-1, and GSK3β phosphorylation | Alzheimer’s disease | [92] |
| S100B | Brain-derived murine primary microglial cells | Stimulates M1 polarization Triggers increases in inducible NO synthase (iNOS), TNF-α, and IL-6 Stimulates decreases in IL-10 and TGF-β production | Cerebral ischemia | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cross, K.; Vetter, S.W.; Alam, Y.; Hasan, M.Z.; Nath, A.D.; Leclerc, E. Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses. Biomolecules 2024, 14, 1550. https://doi.org/10.3390/biom14121550
Cross K, Vetter SW, Alam Y, Hasan MZ, Nath AD, Leclerc E. Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses. Biomolecules. 2024; 14(12):1550. https://doi.org/10.3390/biom14121550
Chicago/Turabian StyleCross, Kaylen, Stefan W. Vetter, Yousuf Alam, Md. Zahidul Hasan, Anupom Deb Nath, and Estelle Leclerc. 2024. "Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses" Biomolecules 14, no. 12: 1550. https://doi.org/10.3390/biom14121550
APA StyleCross, K., Vetter, S. W., Alam, Y., Hasan, M. Z., Nath, A. D., & Leclerc, E. (2024). Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses. Biomolecules, 14(12), 1550. https://doi.org/10.3390/biom14121550