
A Challenging Correlation between Tumor Cellularity and Somatic Variant Allele Fraction in Lung and Colorectal Cancers—Specimens of Low Tumor Percentage Should Be Analyzed with Caution


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. DNA Extraction
2.3. Cancer Hotspot Panel Library Preparation, Sequencing, and Data Analysis
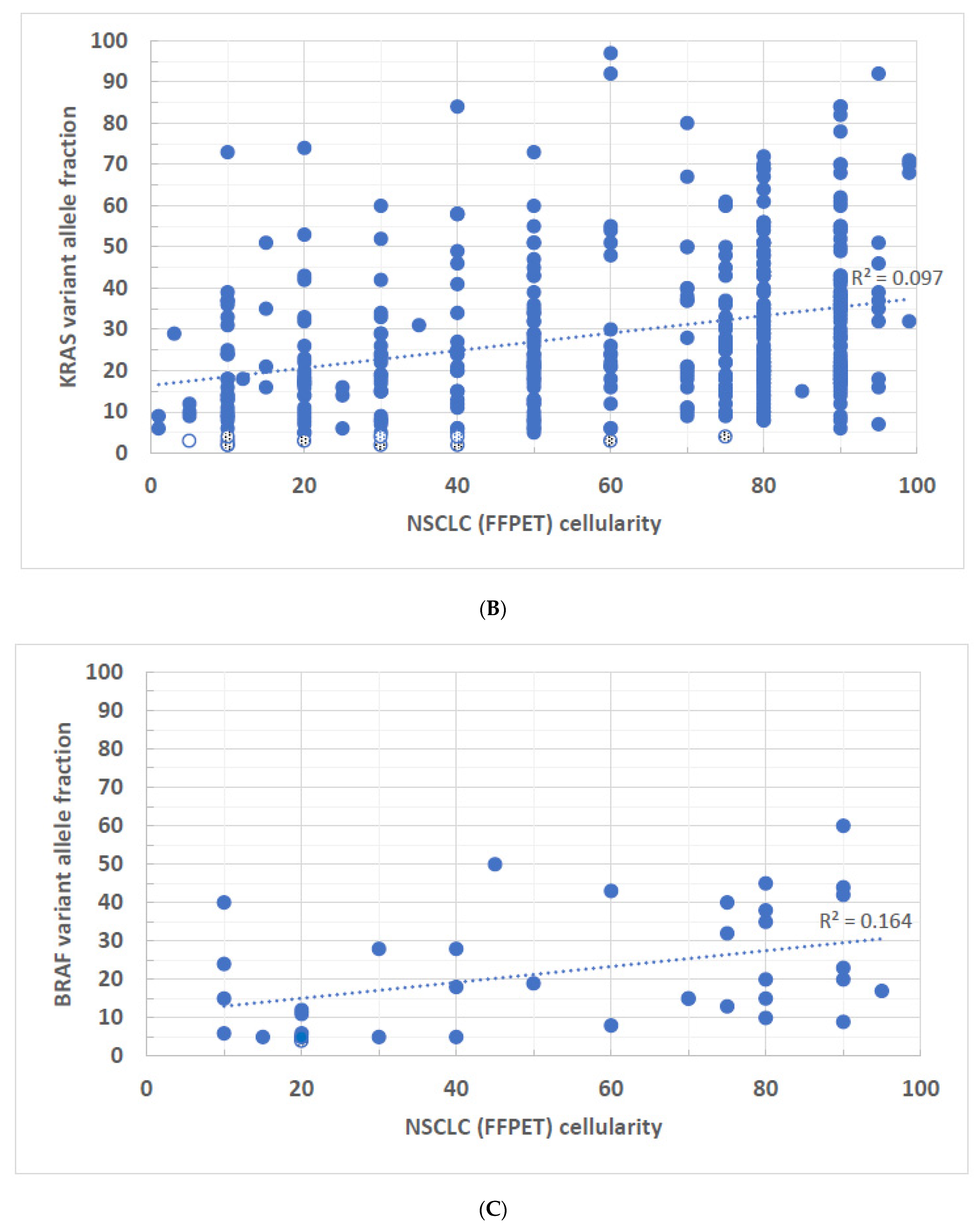
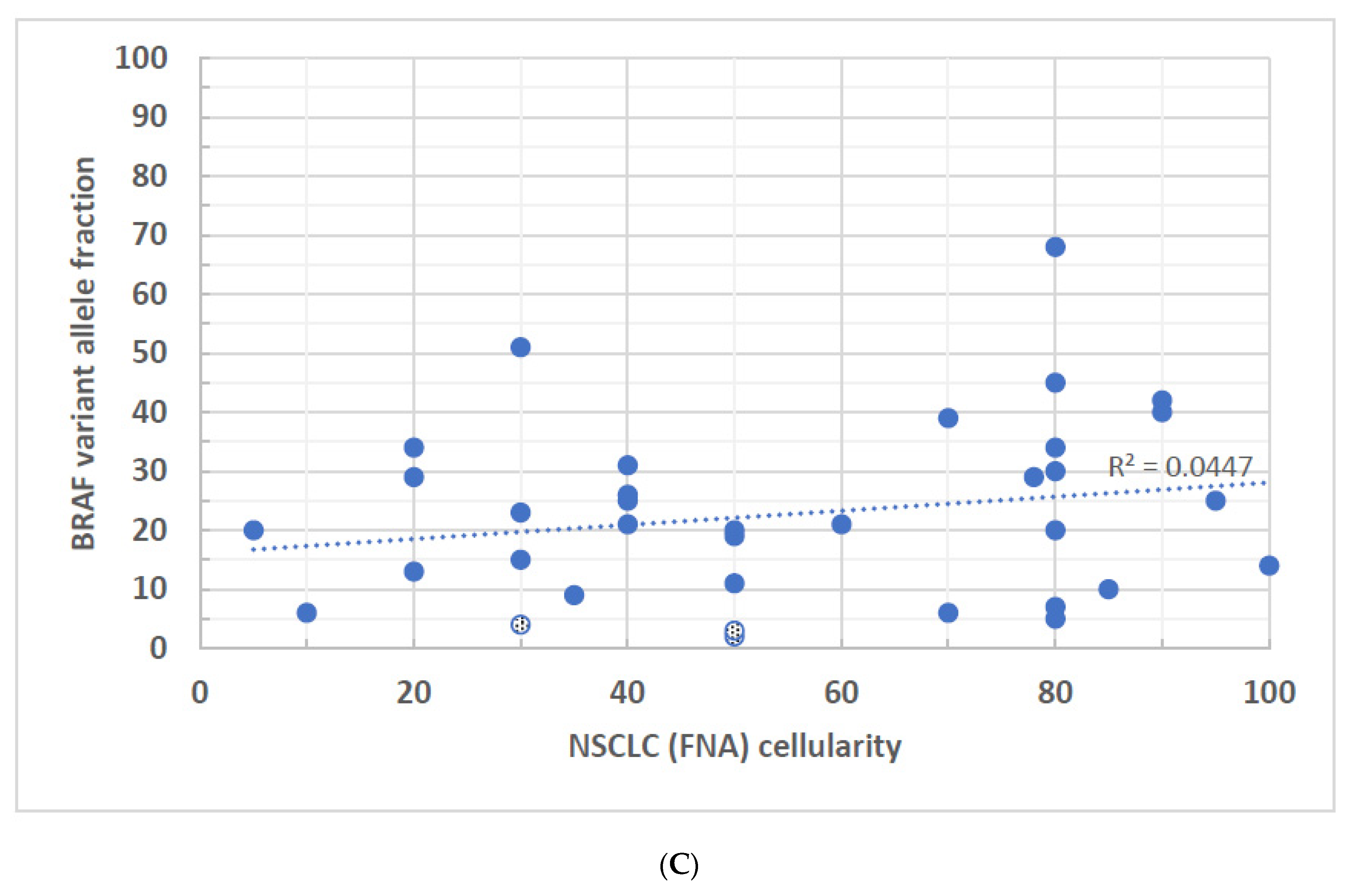
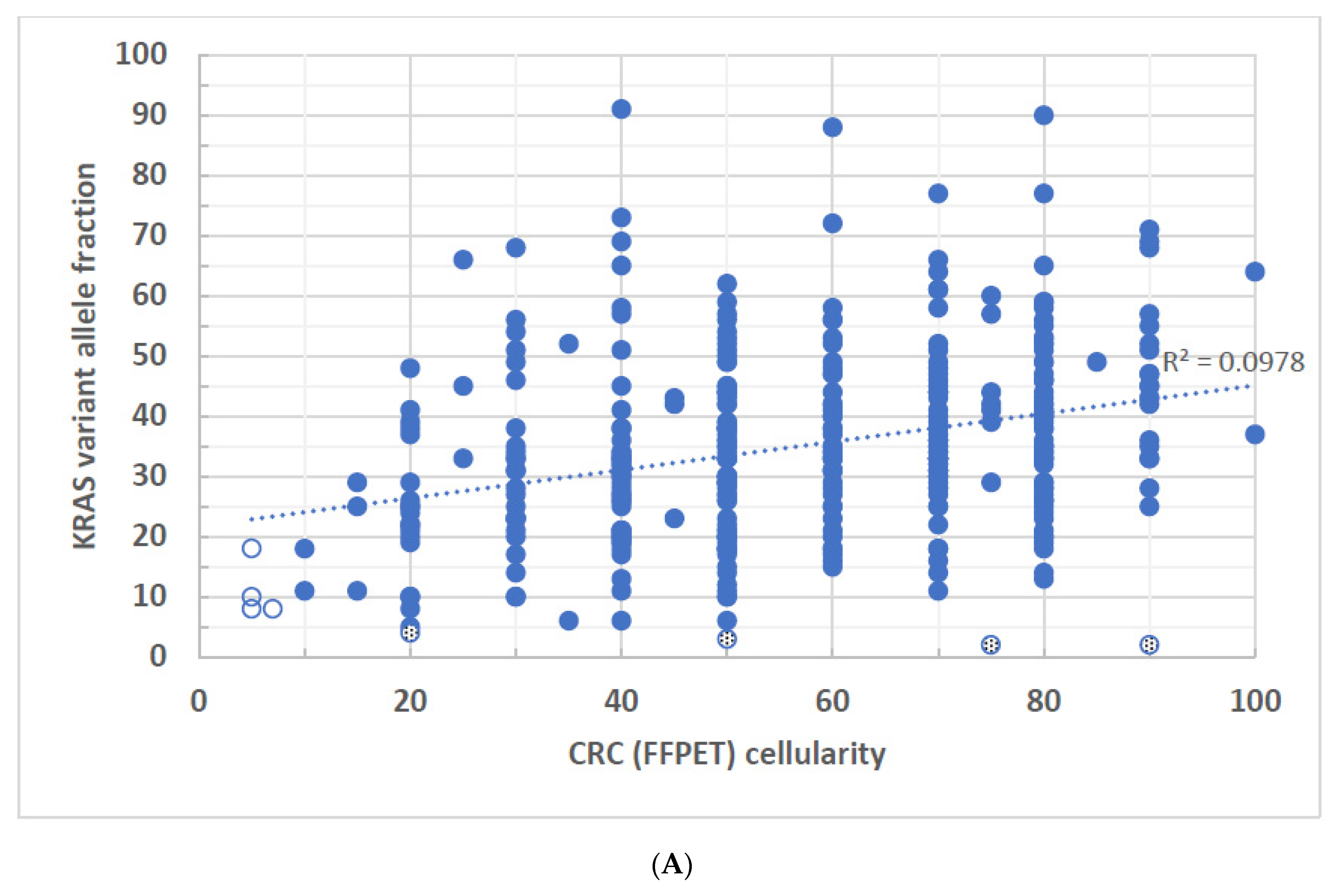
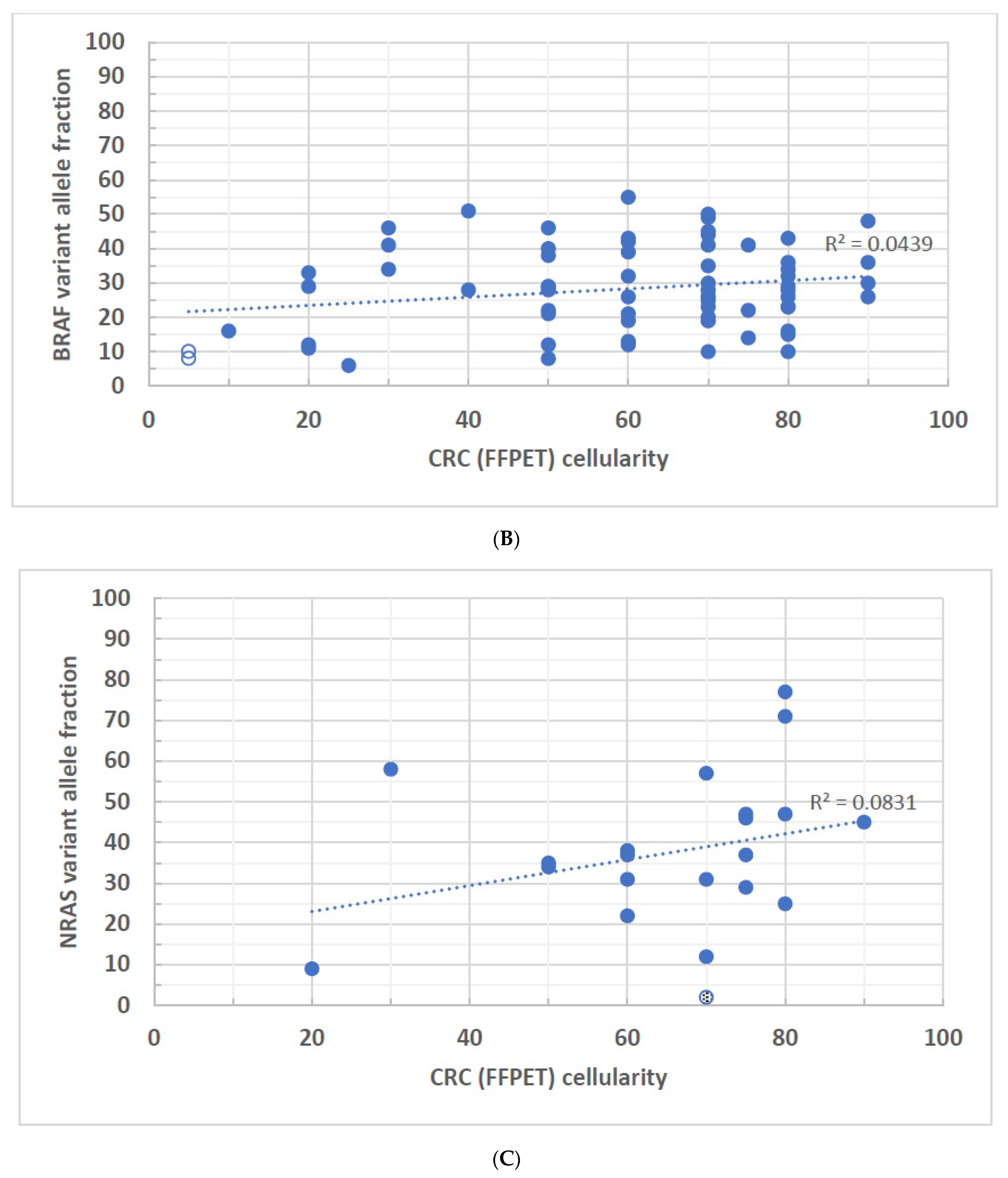
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Thunnissen, E.; Bovee, J.V.; Bruinsma, H.; van den Brule, A.J.; Dinjens, W.; Heideman, D.A.; Meulemans, E.; Nederlof, P.; van Noesel, C.; Prinsen, C.F.; et al. EGFR and KRAS quality assurance schemes in pathology: Generating normative data for molecular predictive marker analysis in targeted therapy. J. Clin. Pathol. 2011, 64, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Warth, A.; Penzel, R.; Brandt, R.; Sers, C.; Fischer, J.R.; Thomas, M.; Herth, F.J.; Dietel, M.; Schirmacher, P.; Blaker, H. Optimized algorithm for Sanger sequencing-based EGFR mutation analyses in NSCLC biopsies. Virchows Arch. 2012, 460, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Pont-Kingdon, G.; Gedge, F.; Wooderchak-Donahue, W.; Schrijver, I.; Weck, K.E.; Kant, J.A.; Oglesbee, D.; Bayrak-Toydemir, P.; Lyon, E.; Biochemical and Molecular Genetic Resource Committee of the College of American Pathologists. Design and analytical validation of clinical DNA sequencing assays. Arch. Pathol. Lab. Med. 2012, 136, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Lamy, A.; Blanchard, F.; Le Pessot, F.; Sesboue, R.; Di Fiore, F.; Bossut, J.; Fiant, E.; Frebourg, T.; Sabourin, J.C. Metastatic colorectal cancer KRAS genotyping in routine practice: Results and pitfalls. Mod. Pathol. 2011, 24, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Viray, H.; Li, K.; Long, T.A.; Vasalos, P.; Bridge, J.A.; Jennings, L.J.; Halling, K.C.; Hameed, M.; Rimm, D.L. A prospective, multi-institutional diagnostic trial to determine pathologist accuracy in estimation of percentage of malignant cells. Arch. Pathol. Lab. Med. 2013, 137, 1545–1549. [Google Scholar] [CrossRef]
- Tembuyser, L.; Ligtenberg, M.J.; Normanno, N.; Delen, S.; van Krieken, J.H.; Dequeker, E.M. Higher quality of molecular testing, an unfulfilled priority: Results from external quality assessment for KRAS mutation testing in colorectal cancer. J. Mol. Diagn. 2014, 16, 371–377. [Google Scholar] [CrossRef]
- Alkorta-Aranburu, G.; Carmody, D.; Cheng, Y.W.; Nelakuditi, V.; Ma, L.; Dickens, J.T.; Das, S.; Greeley, S.A.W.; Del Gaudio, D. Phenotypic heterogeneity in monogenic diabetes: The clinical and diagnostic utility of a gene panel-based next-generation sequencing approach. Mol. Genet. Metab. 2014, 113, 315–320. [Google Scholar] [CrossRef]
- Reynolds, J.P.; Zhou, Y.; Jakubowski, M.A.; Wang, Z.; Brainard, J.A.; Klein, R.D.; Farver, C.F.; Almeida, F.A.; Cheng, Y.W. Next-generation sequencing of liquid-based cytology non-small cell lung cancer samples. Cancer Cytopathol. 2017, 125, 178–187. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Stefaniuk, C.; Jakubowski, M.A. Real-time PCR and targeted next-generation sequencing in the detection of low level EGFR mutations: Instructive case analyses. Respir. Med. Case Rep. 2019, 28, 100901. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- La Fleur, L.; Falk-Sorqvist, E.; Smeds, P.; Berglund, A.; Sundstrom, M.; Mattsson, J.S.; Branden, E.; Koyi, H.; Isaksson, J.; Brunnstrom, H.; et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef]
- Hsu, W.H.; Yang, J.C.; Mok, T.S.; Loong, H.H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29 (Suppl. S1), i3–i9. [Google Scholar] [CrossRef]
- van Eijk, R.; Licht, J.; Schrumpf, M.; Talebian Yazdi, M.; Ruano, D.; Forte, G.I.; Nederlof, P.M.; Veselic, M.; Rabe, K.F.; Annema, J.T.; et al. Rapid KRAS, EGFR, BRAF and PIK3CA mutation analysis of fine needle aspirates from non-small-cell lung cancer using allele-specific qPCR. PLoS ONE 2011, 6, e17791. [Google Scholar] [CrossRef]
- Al-Shamsi, H.O.; Jones, J.; Fahmawi, Y.; Dahbour, I.; Tabash, A.; Abdel-Wahab, R.; Abousamra, A.O.; Shaw, K.R.; Xiao, L.; Hassan, M.M.; et al. Molecular spectrum of KRAS, NRAS, BRAF, PIK3CA, TP53, and APC somatic gene mutations in Arab patients with colorectal cancer: Determination of frequency and distribution pattern. J. Gastrointest. Oncol. 2016, 7, 882–902. [Google Scholar] [CrossRef]
- Bazan, V.; Migliavacca, M.; Zanna, I.; Tubiolo, C.; Grassi, N.; Latteri, M.A.; La Farina, M.; Albanese, I.; Dardanoni, G.; Salerno, S.; et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann. Oncol. 2002, 13, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Sameer, A.S. Colorectal cancer: Molecular mutations and polymorphisms. Front. Oncol. 2013, 3, 114. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.J.; Kummer, J.A.; de Bruin, P.C.; Bol, M.; van den Tweel, J.G.; Seldenrijk, K.A.; Willems, S.M.; Offerhaus, G.J.; de Weger, R.A.; van Diest, P.J.; et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod. Pathol. 2014, 27, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Nones, K.; Miller, D.; Harliwong, I.; Kassahn, K.S.; Pinese, M.; Pajic, M.; Gill, A.J.; Johns, A.L.; Anderson, M.; et al. Qpure: A tool to estimate tumor cellularity from genome-wide single-nucleotide polymorphism profiles. PLoS ONE 2012, 7, e45835. [Google Scholar] [CrossRef] [PubMed]
- Viray, H.; Coulter, M.; Li, K.; Lane, K.; Madan, A.; Mitchell, K.; Schalper, K.; Hoyt, C.; Rimm, D.L. Automated objective determination of percentage of malignant nuclei for mutation testing. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Gold, H.D.; Luquette, L.J.; Park, P.J. Detecting Somatic Mutations in Normal Cells. Trends Genet. 2018, 34, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.S.; Coorens, T.H.H.; Palles, C.; Mitchell, E.; Abascal, F.; Olafsson, S.; Lee, B.C.H.; Lawson, A.R.J.; Lee-Six, H.; Moore, L.; et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat. Genet. 2021, 53, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Portier, B.P.; Kanagal-Shamanna, R.; Luthra, R.; Singh, R.; Routbort, M.J.; Handal, B.; Reddy, N.; Barkoh, B.A.; Zuo, Z.; Medeiros, L.J.; et al. Quantitative assessment of mutant allele burden in solid tumors by semiconductor-based next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 559–572. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | Number | Percentage |
|---|---|---|---|
| EGFR | Ex19 deletion | 73 | 43.5% |
| Ex19 insertion | 3 | 1.8% | |
| Ex20 insertion | 13 | 7.7% | |
| G719A/S | 4 | 2.4% | |
| L747P | 2 | 1.2% | |
| L858R | 61 | 36.3% | |
| L861Q | 4 | 2.4% | |
| M600V | 2 | 1.2% | |
| Other (P733Q, R776C, T751I, V742I, V769L) | 6 | 3.6% | |
| KRAS | G12A/C/D/F/R/S/V | 343 | 84.9% |
| G13C/D/E/F/R/V | 36 | 8.9% | |
| L19F | 2 | 0.5% | |
| A59G/T | 2 | 0.5% | |
| Q61H/L/R | 21 | 5.2% | |
| BRAF | G464V | 2 | 5.4% |
| G466R/V | 3 | 8.1% | |
| G469A/E/R/S/V | 7 | 18.9% | |
| N581I/S | 5 | 13.5% | |
| D594G/N | 3 | 8.1% | |
| V600E/K | 14 | 37.8% | |
| Other (L597R, R462T, p.Thr599_Val600delinsMet) | 3 | 8.1% |
| Gene | Variant | Number | Percentage |
|---|---|---|---|
| EGFR | Ex19 deletion | 59 | 50.9% |
| Ex19 insertion | 1 | 0.9% | |
| Ex20 insertion | 11 | 9.5% | |
| G719A/S | 4 | 3.4% | |
| L858R | 34 | 29.3% | |
| L861Q | 4 | 3.4% | |
| Other (G721A, L747P, R776L) | 3 | 2.6% | |
| KRAS | G12A/C/D/F/R/S/V | 262 | 87.0% |
| G13C/D/R/V | 19 | 6.3% | |
| Q61H/K/L | 18 | 6.0% | |
| A146T/V | 2 | 0.7% | |
| BRAF | G464V | 1 | 3.0% |
| G466A/V | 5 | 15.2% | |
| G469A/R/V | 9 | 27.3% | |
| N581I/S | 3 | 9.1% | |
| N594G/N | 5 | 15.2% | |
| K601E | 2 | 6.1% | |
| V600E | 8 | 24.2% |
| Gene | Variant | Number | Percentage |
|---|---|---|---|
| KRAS | G12A/C/D/F/R/S/V | 243 | 66.9% |
| G13C/D/R | 68 | 18.7% | |
| Q22K | 2 | 0.6% | |
| A59T | 2 | 0.6% | |
| Q61H/K/L/R | 20 | 5.5% | |
| K117N | 3 | 0.8% | |
| A146R | 1 | 0.3% | |
| A146T | 24 | 6.6% | |
| BRAF | D594G | 3 | 4.5% |
| V600E | 57 | 86.4% | |
| Other (G466R, F468L, G469A, D581I, G596R, K601N) | 6 | 9.1% | |
| NRAS | G12A/C/D/S | 6 | 26.1% |
| G13R | 2 | 8.7% | |
| Q61K/L/R | 15 | 65.2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarabi, S.K.; Zhai, L.; Cheng, Y.-W. A Challenging Correlation between Tumor Cellularity and Somatic Variant Allele Fraction in Lung and Colorectal Cancers—Specimens of Low Tumor Percentage Should Be Analyzed with Caution. Biomolecules 2024, 14, 168. https://doi.org/10.3390/biom14020168
Zarabi SK, Zhai L, Cheng Y-W. A Challenging Correlation between Tumor Cellularity and Somatic Variant Allele Fraction in Lung and Colorectal Cancers—Specimens of Low Tumor Percentage Should Be Analyzed with Caution. Biomolecules. 2024; 14(2):168. https://doi.org/10.3390/biom14020168
Chicago/Turabian StyleZarabi, Samaneh K., Lidong Zhai, and Yu-Wei Cheng. 2024. "A Challenging Correlation between Tumor Cellularity and Somatic Variant Allele Fraction in Lung and Colorectal Cancers—Specimens of Low Tumor Percentage Should Be Analyzed with Caution" Biomolecules 14, no. 2: 168. https://doi.org/10.3390/biom14020168
APA StyleZarabi, S. K., Zhai, L., & Cheng, Y.-W. (2024). A Challenging Correlation between Tumor Cellularity and Somatic Variant Allele Fraction in Lung and Colorectal Cancers—Specimens of Low Tumor Percentage Should Be Analyzed with Caution. Biomolecules, 14(2), 168. https://doi.org/10.3390/biom14020168