
The Role of Lung Microbiome in Fibrotic Interstitial Lung Disease—A Systematic Review
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
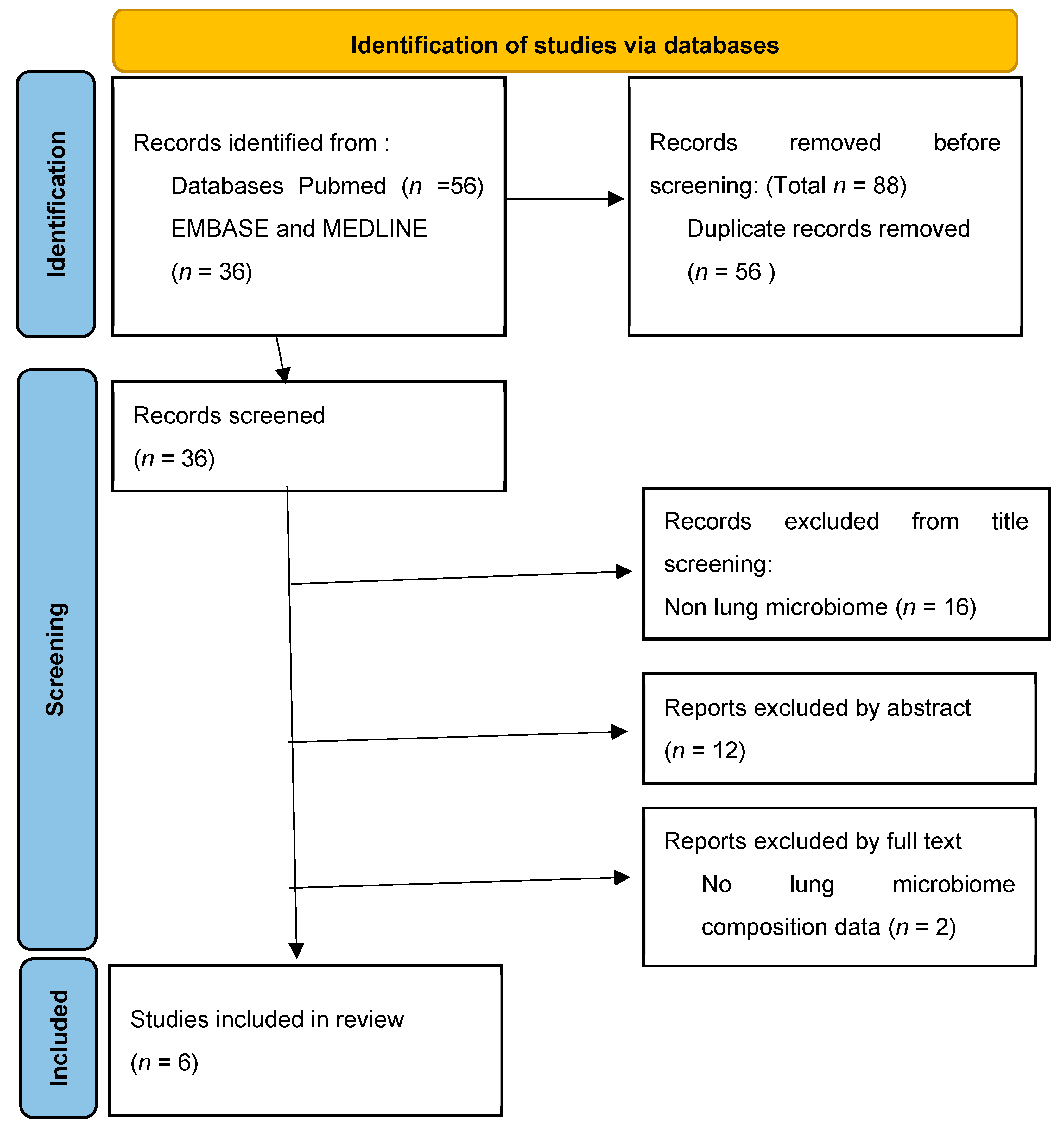
2.1. Search Strategy
2.2. Study Selection
3. Results
3.1. Lung Microbiome in Idiopathic Pulmonary Fibrosis
3.1.1. Lung Microbiome in Stable IPF
3.1.2. Lung Microbiome in Acute Exacerbation of IPF (AE-IPF)
3.2. Lung Microbiome in Other Interstitial Lung Disease
3.2.1. Lung Microbiome in Hypersensitivity Pneumonitis
3.2.2. Lung Microbiome in Sarcoidosis
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; King, T.E., Jr.; Raghu, G. Diffuse parenchymal lung disease. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 787–796. [Google Scholar]
- Fischer, A.; Antoniou, K.M.; Brown, K.K.; Cadranel, J.; Corte, T.J.; du Bois, R.M.; Lee, J.S.; Leslie, K.O.; Lynch, D.A.; Matteson, E.L.; et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015, 46, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Salton, F.; Ruaro, B.; Confalonieri, P.; Confalonieri, M. Epithelial–Mesenchymal Transition: A Major Pathogenic Driver in Idiopathic Pulmonary Fibrosis? Medicina 2020, 56, 608. [Google Scholar] [CrossRef]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic Pulmonary Fibrosis Clinical Research Network. Interstitial lung disease: Diagnostic evaluation and management. BMJ 2019, 367, l5378. [Google Scholar]
- Axelsson, G.T.; Gudmundsson, G.; Pratte, K.A.; Aspelund, T.; Putman, R.K.; Sanders, J.L.; Gudmundsson, E.F.; Hatabu, H.; Gudmundsdottir, V.; Gudjonsson, A.; et al. The Proteomic Profile of Interstitial Lung Abnormalities. Am. J. Respir. Crit. Care Med. 2022, 206, 337–346. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Trachalaki, A.; Sultana, N.; Wells, A.U. An update on current and emerging drug treatments for idiopathic pulmonary fibrosis. Expert Opin. Pharmacother. 2023, 24, 1125–1142. [Google Scholar] [CrossRef]
- Vacchi, C.; Sebastiani, M.; Cassone, G.; Cerri, S.; Della Casa, G.; Salvarani, C.; Manfredi, A. Therapeutic Options for the Treatment of Interstitial Lung Disease Related to Connective Tissue Diseases. A Narrative Review. J. Clin. Med. 2020, 9, 407. [Google Scholar] [CrossRef]
- Hambly, N.; Farooqi, M.M.; Dvorkin-Gheva, A.; Donohoe, K.; Garlick, K.; Scallan, C.; Chong, S.G.; MacIsaac, S.; Assayag, D.; Johannson, K.A.; et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur. Respir. J. 2022, 60, 2102571. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Kreuter, M.; Olson, A.; Fischer, A.; Bendstrup, E.; Wells, C.D.; Denton, C.P.; Mounir, B.; Zouad-Lejour, L.; Quaresma, M.; et al. Progressive fibrosing interstitial lung diseases: Current practice in diagnosis and management. Curr. Med. Res. Opin. 2019, 35, 2015–2024. [Google Scholar] [CrossRef]
- Nasser, M.; Larrieu, S.; Si-Mohamed, S.; Ahmad, K.; Boussel, L.; Brevet, M.; Chalabreysse, L.; Fabre, C.; Marque, S.; Revel, D.; et al. Progressive fibrosing interstitial lung disease: A clinical cohort (the PROGRESS study). Eur. Respir. J. 2020, 57, 2002718. [Google Scholar] [CrossRef]
- Zaibi, H.; Chaabi, K.; Ben Jemia, E.; Khalfallah, N.; Ben Amar, J.; Aouina, H. Survival and mortality associated factors in progressive fibrosing interstitial disease. Eur. Respir. J. 2022, 60 (Suppl. S66), 4062. [Google Scholar]
- Nasser, M.; Larrieu, S.; Boussel, L.; Si-Mohamed, S.; Diaz, F.; Marque, S.; Massol, J.; Revel, D.; Thivolet-Bejui, F.; Chalabreysse, L.; et al. Epidemiology and mortality of non-idiopathic pulmonary fibrosis (IPF) progressive fibrosing interstitial lung disease (PF-ILD) using the French national health insurance system (SNDS) database in France: The PROGRESS study. Eur. Respir. J. 2020, 56, 444. [Google Scholar]
- Chen, Y.-H.; Lee, T.-J.; Hsieh, H.-J.; Hsieh, S.-C.; Wang, H.-C.; Chang, Y.-C.; Yu, C.-J.; Chien, J.-Y. Clinical outcomes and risk factors of progressive pulmonary fibrosis in primary Sjögren’s syndrome-associated interstitial lung disease. BMC Pulm. Med. 2023, 23, 268. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, M.F.; Cookson, W.O. The lung microbiome in health and disease. Clin. Med. 2017, 17, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Faner, R.; Sibila, O.; Agustí, A.; Bernasconi, E.; Chalmers, J.D.; Huffnagle, G.B.; Manichanh, C.; Molyneaux, P.L.; Paredes, R.; Brocal, V.P.; et al. The microbiome in respiratory medicine: Current challenges and future perspectives. Eur. Respir. J. 2017, 49, 1602086. [Google Scholar] [CrossRef] [PubMed]
- Carney, S.M.; Clemente, J.C.; Cox, M.J.; Dickson, R.P.; Huang, Y.J.; Kitsios, G.D.; Kloepfer, K.M.; Leung, J.M.; LeVan, T.D.; Molyneaux, P.L.; et al. Methods in Lung Microbiome Research. Am. J. Respir. Cell Mol. Biol. 2020, 62, 283–299. [Google Scholar] [CrossRef]
- Cox, M.J.; Ege, M.J.; von Mutius, E. The Lung Microbiome; European Respiratory Society (ERS): Sheffield, UK, 2019; ISBN 9781849841016. [Google Scholar] [CrossRef]
- Baker, J.M.; Hinkle, K.J.; McDonald, R.A.; Brown, C.A.; Falkowski, N.R.; Huffnagle, G.B.; Dickson, R.P. Whole lung tissue is the preferred sampling method for amplicon-based characterization of murine lung microbiota. Microbiome 2021, 9, 99. [Google Scholar] [CrossRef]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e8578. [Google Scholar] [CrossRef]
- Morris, A.; Beck, J.M.; Schloss, P.D.; Campbell, T.B.; Crothers, K.; Curtis, J.L.; Flores, S.C.; Fontenot, A.P.; Ghedin, E.; Huang, L.; et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 2013, 187, 1067–1075. [Google Scholar] [CrossRef]
- Huang, Y.J.; Boushey, H.A. The microbiome in asthma. J. Allergy Clin. Immunol. 2014, 135, 25–30. [Google Scholar] [CrossRef]
- Huang, Y.J.; Kim, E.; Cox, M.J.; Brodie, E.L.; Brown, R.; Wiener-Kronish, J.P.; Lynch, S.V. A Persistent and Diverse Airway Microbiota Present during Chronic Obstructive Pulmonary Disease Exacerbations. OMICS J. Integr. Biol. 2010, 14, 9–59. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.G.; Mallia, P.; Russell, K.E.; Russell, A.-M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Falkowski, N.R.; Huffnagle, G.B.; Curtis, J.L. Bacterial Topography of the Healthy Human Lower Respiratory Tract. mBio 2017, 8, e02287-16. [Google Scholar] [CrossRef] [PubMed]
- Sze, M.A.; Schloss, P.D. Leveraging Existing 16S rRNA Gene Surveys to Identify Reproducible Biomarkers in Individuals with Colorectal Tumors. mBio 2018, 9, e00630-18, Erratum in mBio 2018, 9, e02076-18. [Google Scholar] [CrossRef]
- Man, W.H.; De Steenhuijsen Piters, W.A.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, 71. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Current and future treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Man, M.A.; Ungur, R.A.; Motoc, N.S.; Pop, L.A.; Berindan-Neagoe, I.; Ruta, V.M. Lung Microbiota in Idiopathic Pulmonary Fibrosis, Hypersensitivity Pneumonitis, and Unclassified Interstitial Lung Diseases: A Preliminary Pilot Study. Diagnostics 2023, 13, 3157. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, S.-F.; Espindola, M.S.; Vij, R.; Oldham, J.M.; Huffnagle, G.B.; Erb-Downward, J.R.; Flaherty, K.R.; Moore, B.B.; White, E.S.; et al. Microbes Are Associated with Host Innate Immune Response in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 208–219. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Ashley, S.L.; Gurczynski, S.J.; Xia, M.; Wilke, C.; Falkowski, N.R.; Norman, K.C.; Arnold, K.B.; Huffnagle, G.B.; Salisbury, M.L.; et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1127–1138. [Google Scholar] [CrossRef]
- Kitsios, G.D.; Rojas, M.; Kass, D.J.; Fitch, A.; Sembrat, J.C.; Qin, S.; Veraldi, K.L.; Gibson, K.F.; Lindell, K.; Pilewski, J.M.; et al. Microbiome in lung explants of idiopathic pulmonary fibrosis: A case–control study in patients with end-stage fibrosis. Thorax 2017, 73, 481–484. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef]
- Weng, D.; Chen, X.-Q.; Qiu, H.; Zhang, Y.; Li, Q.-H.; Zhao, M.-M.; Wu, Q.; Chen, T.; Hu, Y.; Wang, L.-S.; et al. The Role of Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2019, 2019, 5160694. [Google Scholar] [CrossRef]
- Ohshimo, S.; Ishikawa, N.; Horimasu, Y.; Hattori, N.; Hirohashi, N.; Tanigawa, K.; Costabel, U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir. Med. 2014, 108, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.C.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef]
- Salisbury, M.L.; Myers, J.L.; Belloli, E.A.; Kazerooni, E.A.; Martinez, F.J.; Flaherty, K.R. Diagnosis and Treatment of Fibrotic Hypersensitivity Pneumonia. Where We Stand and Where We Need to Go. Am. J. Respir. Crit. Care Med. 2017, 196, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Varone, F.; Iovene, B.; Sgalla, G.; Calvello, M.; Calabrese, A.; Larici, A.R.; Richeldi, L. Fibrotic Hypersensitivity Pneumonitis: Diagnosis and Management. Lung 2020, 198, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Churg, A. Hypersensitivity pneumonitis: New concepts and classifications. Mod. Pathol. 2022, 35 (Suppl. S1), 15–27. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, R.; Wu, B.G.; Barnett, J.; Ghai, P.; Kingston, S.; Hewitt, R.J.; Feary, J.; Li, Y.; Chua, F.; Wu, Z.; et al. The Respiratory Microbiome in Chronic Hypersensitivity Pneumonitis Is Distinct from That of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Bernardinello, N.; Petrarulo, S.; Balestro, E.; Cocconcelli, E.; Veltkamp, M.; Spagnolo, P. Pulmonary Sarcoidosis: Diagnosis and Differential Diagnosis. Diagnostics 2021, 11, 1558. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Birnie, D.; Dwivedi, G. Current perspectives on the immunopathogenesis of sarcoidosis. Respir. Med. 2020, 173, 106161. [Google Scholar] [CrossRef]
- Inaoka, P.T.; Shono, M.; Kamada, M.; Espinoza, J.L. Host-microbe interactions in the pathogenesis and clinical course of sarcoidosis. J. Biomed. Sci. 2019, 26, 45. [Google Scholar] [CrossRef]
- Zimmermann, A.; Knecht, H.; Häsler, R.; Zissel, G.; Gaede, K.I.; Hofmann, S.; Nebel, A.; Müller-Quernheim, J.; Schreiber, S.; Fischer, A. Atopobium and Fusobacterium as novel candidates for sarcoidosis-associated microbiota. Eur. Respir. J. 2017, 50, 1600746. [Google Scholar] [CrossRef]
- Clarke, E.L.; Lauder, A.P.; Hofstaedter, C.E.; Hwang, Y.; Fitzgerald, A.S.; Imai, I.; Biernat, W.; Rękawiecki, B.; Majewska, H.; Dubaniewicz, A.; et al. Microbial Lineages in Sarcoidosis. A Metagenomic Analysis Tailored for Low–Microbial Content Samples. Am. J. Respir. Crit. Care Med. 2018, 197, 225–234. [Google Scholar] [CrossRef]
- Knudsen, K.S.; Lehmann, S.; Nielsen, R.; Tangedal, S.; Paytuvi-Gallart, A.; Sanseverino, W.; Martinsen, E.M.H.; Hiemstra, P.S.; Eagan, T.M. The lower airways microbiota and antimicrobial peptides indicate dysbiosis in sarcoidosis. Microbiome 2022, 10, 175. [Google Scholar] [CrossRef]
- Rajan, S.K.; Cottin, V.; Dhar, R.; Danoff, S.; Flaherty, K.R.; Brown, K.K.; Mohan, A.; Renzoni, E.; Mohan, M.; Udwadia, Z.; et al. Progressive pulmonary fibrosis: An expert group consensus statement. Eur. Respir. J. 2023, 61, 2103187. [Google Scholar] [CrossRef]
- Sikora, M.; Jastrzębski, D.; Pilzak, K.; Ziora, D.; Hall, B.; Żebrowska, A. Impact of physical functional capacity on quality of life in patients with interstitial lung diseases. Respir. Physiol. Neurobiol. 2023, 313, 104064. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Z.; Ma, R.; Wang, J.; Wu, N.; Fan, Y.; Ye, Q. Prognostic Predictive Characteristics in Patients With Fibrosing Interstitial Lung Disease: A Retrospective Cohort Study. Front. Pharmacol. 2022, 13, 924754. [Google Scholar] [CrossRef]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Ichikado, K.; Yasuda, Y.; Anan, K.; Suga, M. Azithromycin for idiopathic acute exacerbation of idiopathic pulmonary fibrosis: A retrospective single-center study. BMC Pulm. Med. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bhattacharya, P.; Paul, S.; Paul, R.; Swarnakar, S. An alternative therapy for idiopathic pulmonary fibrosis by doxycycline through matrix metalloproteinase inhibition. Lung 2011, 28, 174–179. [Google Scholar] [CrossRef]
- Varney, V.; Parnell, H.; Salisbury, D.; Ratnatheepan, S.; Tayar, R. A double blind randomised placebo controlled pilot study of oral co-trimoxazole in advanced fibrotic lung disease. Pulm. Pharmacol. Ther. 2008, 21, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.T.; Marsland, B.J. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol. 2019, 12, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, K.; Alrawi, Z.; Persson, A.; Jönsson, G.; Marsal, J. Intestinal dysbiosis is common in systemic sclerosis and associated with gastrointestinal and extraintestinal features of disease. Arthritis Res. 2016, 18, 278. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.L.; Gold, M.J.; Reynolds, L.A.; Willing, B.P.; Dimitriu, P.; Thorson, L.; Redpath, S.A.; Perona-Wright, G.; Blanchet, M.-R.; Mohn, W.W.; et al. Perinatal antibiotic-induced shifts in gut microbiota have differential effects on inflammatory lung diseases. J. Allergy Clin. Immunol. 2014, 135, 100–109.e5. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Term | Definition |
|---|---|
| Microbiome | community of microorganisms, including commensal, symbiotic, and pathogenic ones, found in a particular environment or body space |
| Microbiota | collection of living microorganisms present in a certain environment |
| Metagenome | genetic information of the microbiota obtained through genetic sequencing, which is then analyzed, organized, and identified through computational tools with the help of previously known sequence databases’ |
| 16S rRNA gene | component of the 30S small subunit of prokaryotic ribosomes, used in molecular studies due to its extremely slow rate of evolution and the presence of both variable and constant regions; |
| Operational taxonomic unit (OTU) | clusters of similar 16S rRNA gene sequences, with each OTU representing a taxonomic unit of a bacteria family or genus, depending on the sequence similarity threshold; |
| Dysbiosis | an imbalance in the composition of the microbiota of a given niche, related to changes in local conditions; |
| Abundance | total number of bacteria individuals in a specific sample; |
| Richness | is the number of different species/OTUs in a specific sample; |
| α-diversity | a measure of diversity within a sample and is based on the relative abundance of taxa; |
| β-diversity | a measure of the differences between samples from different groups; |
| Author and Year | Design of the Study | Sample Size | Microbiome Assessment | Sample | Main Findings |
|---|---|---|---|---|---|
| Molyneaux et. al. 2014 [29] | Monocenter, observational, longitudinal, prospective | IPF patients:65 COPD patients: 17 Healthy controls: 27 | PCR amplification of the 16S rRNA gene | BAL from middle lobe or lingular segment | Increased bacterial load was correlated with a more rapid progression and mortality rate in IPF. A greater bacterial load was associated with the presence of the MUC5B s3570590 T allele. Streptococcus spp. Or Staphylococcus spp. In the composition of lung microbiome was strongly associated with disease progression. |
| Han et. al. 2017 [36] | Multicenter, Observational, Longitudinal, prospective | IPF patients: 55 | DNA sequencing | BAL from middle lobe, Lung tissues from explants; | Streptococcus and Staphylococcus OTUs appear to be associated with higher risk of disease progression; |
| Huang et. al. 2017 [38] | Multicenter, Observational prospective | IPF patients: 68 | PCR amplification of the 16S rRNA gene | BAL from middle lobe | Prevotella and Staphylococcus are negatively correlated with inflammatory responses; |
| O’Dwyer et al. 2019 [39] | Multicenter, Observational, prospective | IPF patients: 68 | ddPCR (droplet digital PCR) for 16S rRNA gene | BAL from middle lobe | Greater bacterial burden is associated with progression in IPF and lung dysbiosis was correlated with increased inflammation, pro-fibrotic signaling and faulty repair; |
| Kitosis et al. 2018 [40] | Case-control | IPF patients end stage: 40 Controls: 37 | PCR amplification of the 16S rRNA gene | Subpleural lung tissue samples with advanced honey combing | Low bacterial signal in the end stage lung similar to controls; |
| Molyneaux et al. 2017 [44] | Monocenter, Observational, prospective | AE-IPF patients: 20 Stable IPD pateints: 15 | PCR amplification of the 16S rRNA gene | BAL from middle lobe | A higher load of bacteria (up to four times) carried AE-IPF patients than the stable IPF counterparts; |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puiu, R.; Motoc, N.S.; Lucaciu, S.; Ruta, M.V.; Rajnoveanu, R.-M.; Todea, D.A.; Man, M.A. The Role of Lung Microbiome in Fibrotic Interstitial Lung Disease—A Systematic Review. Biomolecules 2024, 14, 247. https://doi.org/10.3390/biom14030247
Puiu R, Motoc NS, Lucaciu S, Ruta MV, Rajnoveanu R-M, Todea DA, Man MA. The Role of Lung Microbiome in Fibrotic Interstitial Lung Disease—A Systematic Review. Biomolecules. 2024; 14(3):247. https://doi.org/10.3390/biom14030247
Chicago/Turabian StylePuiu, Ruxandra, Nicoleta Stefania Motoc, Sergiu Lucaciu, Maria Victoria Ruta, Ruxandra-Mioara Rajnoveanu, Doina Adina Todea, and Milena Adina Man. 2024. "The Role of Lung Microbiome in Fibrotic Interstitial Lung Disease—A Systematic Review" Biomolecules 14, no. 3: 247. https://doi.org/10.3390/biom14030247