
Restoring Mitochondrial Function and Muscle Satellite Cell Signaling: Remedies against Age-Related Sarcopenia
 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ultrastructural Changes in the Aged Skeletal Muscle
3. Age-Related Muscle Changes: An Immunological Perspective
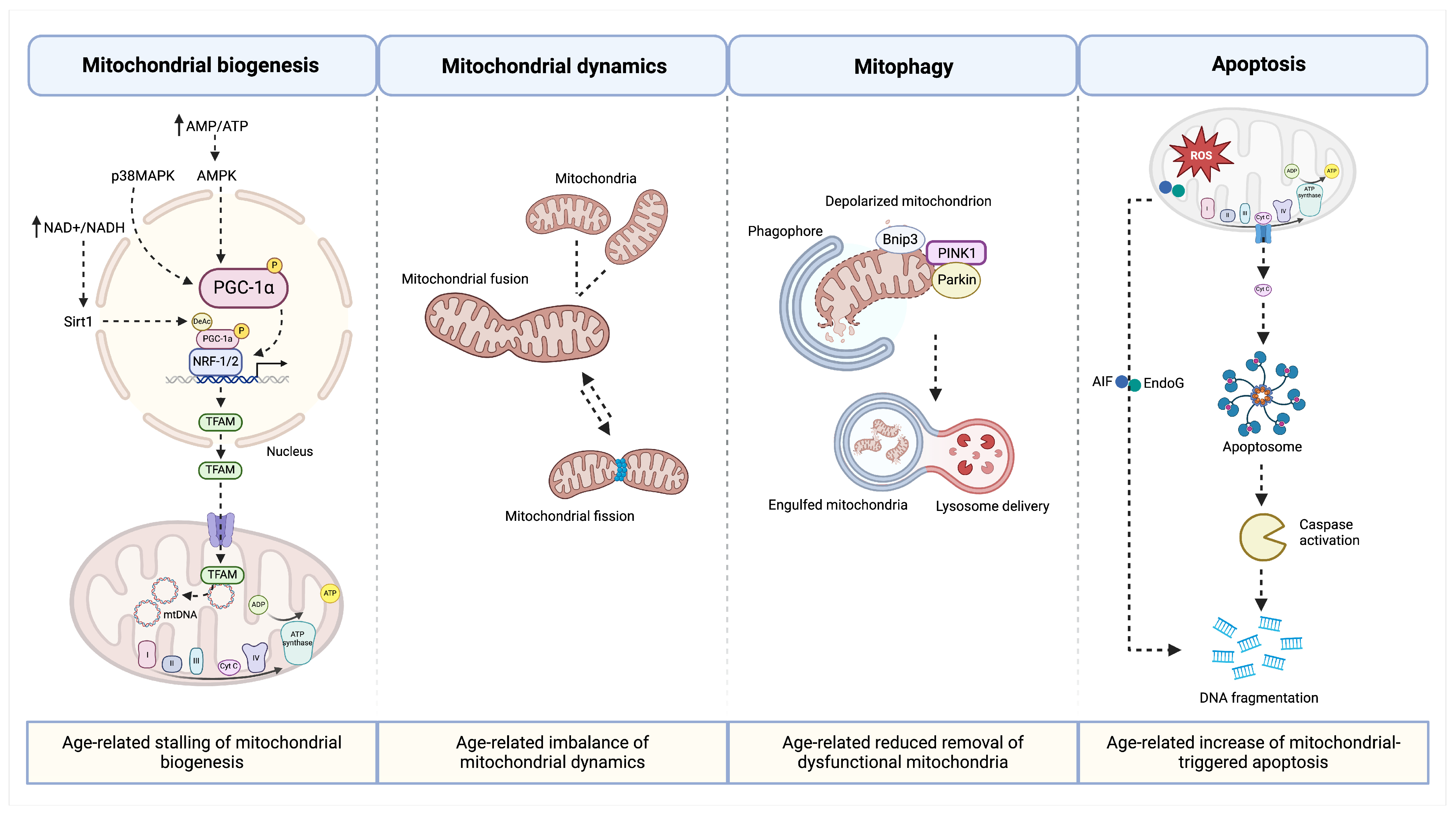
4. Age-Related Changes of Mitochondria in the Muscle
5. Mitochondrial Delivery as a Remedy in Muscle Regeneration
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Bahat, G.G.; Bauer, J.J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 601. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.R.; Dunne, R.F.; Giri, S.; Shachar, S.S.; Caan, B.J. Sarcopenia in the older adult with cancer. J. Clin. Oncol. 2021, 39, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Mesinovic, J.; Zengin, A.; De Courten, B.; Ebeling, P.R.; Scott, D. Sarcopenia and type 2 diabetes mellitus: A bidirectional relationship. Diabetes Metab. Syndr. Obes. 2019, 12, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.; Springer, J.-I.; Anker, S.D. Muscle wasting and sarcopenia in heart failure and beyond: Update 2017. ESC Heart Fail. 2017, 4, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Coenen-Schimke, J.M.; Rys, P.; Nair, K.S. Changes in myosin heavy chain mRNA and protein expression in human skeletal muscle with age and endurance exercise training. J. Appl. Physiol. 2005, 99, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, L.B.; Snijders, T.; Drost, M.; Delhaas, T.; Kadi, F.; Van Loon, L.J.C. Satellite cells in human skeletal muscle; from birth to old age. Age 2014, 36, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Janssen, I.; Heymsfield, S.B.; Ross, R. Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J. Am. Geriatr. Soc. 2002, 50, 889–896. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Picca, A. Mitochondrial quantity and quality in age-related sarcopenia. Int. J. Mol. Sci. 2024, 25, 2052. [Google Scholar] [CrossRef] [PubMed]
- Sirago, G.; Picca, A.; Calvani, R.; Coelho-Júnior, H.J.; Marzetti, E. Mammalian target of rapamycin (mTOR) signaling at the crossroad of muscle fiber fate in sarcopenia. Int. J. Mol. Sci. 2022, 23, 13823. [Google Scholar] [CrossRef]
- Farup, J.; Madaro, L.; Puri, P.L.; Mikkelsen, U.R. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 2015, 6, e1830. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Bossola, M.; Allocca, E.; Menghi, A.; Pesce, V.; Lezza, A.M.S.; Bernabei, R.; Landi, F.; Marzetti, E. Update on mitochondria and muscle aging: All wrong roads lead to sarcopenia. Biol. Chem. 2018, 399, 421–436. [Google Scholar] [CrossRef]
- Brooks, M.J.; Hajira, A.; Mohamed, J.S.; Alway, S.E. Voluntary wheel running increases satellite cell abundance and improves recovery from disuse in gastrocnemius muscles from mice. J. Appl. Physiol. 2018, 124, 1616–1628. [Google Scholar] [CrossRef]
- Picca, A.; Lozanoska-Ochser, B.; Calvani, R.; Coelho-Júnior, H.J.; Leewenburgh, C.; Marzetti, E. Inflammatory, mitochondrial, and senescence-related markers: Underlying biological pathways of muscle aging and new therapeutic targets. Exp. Gerontol. 2023, 178, 112204. [Google Scholar] [CrossRef]
- Myers, M.J.; Shepherd, D.L.; Durr, A.J.; Stanton, D.S.; Mohamed, J.S.; Hollander, J.M.; Alway, S.E. The role of SIRT1 in skeletal muscle function and repair of older mice. J. Cachexia Sarcopenia Muscle 2019, 10, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Ali Pour, P.; Kenney, M.C.; Kheradvar, A. Bioenergetics consequences of mitochondrial transplantation in cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e014501. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef]
- Sasaki, D.; Abe, J.; Takeda, A.; Harashima, H.; Yamada, Y. Transplantation of MITO cells, mitochondria activated cardiac progenitor cells, to the ischemic myocardium of mouse enhances the therapeutic effect. Sci. Rep. 2022, 12, 4344. [Google Scholar] [CrossRef]
- Alway, S.E.; Paez, H.G.; Pitzer, C.R.; Ferrandi, P.J.; Khan, M.M.; Mohamed, J.S.; Carson, J.A.; Deschenes, M.R. Mitochondria transplant therapy improves regeneration and restoration of injured skeletal muscle. J. Cachexia Sarcopenia Muscle 2023, 14, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Edström, L.; Larsson, L. Effects of age on contractile and enzyme-histochemical properties of fast- and slow-twitch single motor units in the rat. J. Physiol. 1987, 392, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Edstrom, L.; Lindegren, B.; Gorza, L.; Schiaffino, S. MHC composition and enzyme-histochemical and physiological properties of a novel fast-twitch motor unit type. Am. J. Physiol. Cell Physiol. 1991, 261, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Biral, D.; Campione, M.; Schiaffino, S. An age-related type IIB to IIX myosin heavy chain switching in rat skeletal muscle. Acta Physiol. Scand. 1993, 147, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Ansved, T.; Edström, L.; Gorza, L.; Schiaffino, S. Effects of age on physiological, immunohistochemical and biochemical properties of fast-twitch single motor units in the rat. J. Physiol. 1991, 443, 257–275. [Google Scholar] [CrossRef]
- Larsson, L.; Ansved, T. Effects of ageing on the motor unit. Prog. Neurobiol. 1995, 45, 397–458. [Google Scholar] [CrossRef] [PubMed]
- De Waard, M.C.; Van Der Pluijm, I.; Zuiderveen Borgesius, N.; Comley, L.H.; Haasdijk, E.D.; Rijksen, Y.; Ridwan, Y.; Zondag, G.; Hoeijmakers, J.H.J.; Elgersma, Y.; et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010, 120, 461–475. [Google Scholar] [CrossRef]
- Shinpo, K.; Kikuchi, S.; Sasaki, H.; Ogata, A.; Moriwaka, F.; Tashiro, K. Selective vulnerability of spinal motor neurons to reactive dicarbonyl compounds, intermediate products of glycation, in vitro: Implication of inefficient glutathione system in spinal motor neurons. Brain Res. 2000, 861, 151–159. [Google Scholar] [CrossRef]
- Punga, A.R.; Ruegg, M.A. Signaling and aging at the neuromuscular synapse: Lessons learnt from neuromuscular diseases. Curr. Opin. Pharmacol. 2012, 12, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; de Cabo, R.; Studenski, S.A.; Ferrucci, L. The neuromuscular junction: Aging at the crossroad between nerves and muscle. Front. Aging Neurosci. 2014, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, Y.I.; Thompson, W.J. Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J. Neurosci. 2011, 31, 14910–14999. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Mimori, Y.; Tanaka, E.; Kohriyama, T.; Nakamura, S. Age-related change in peripheral nerve conduction: Compound muscle action potential duration and dispersion. Gerontology 1999, 45, 168–173. [Google Scholar] [CrossRef] [PubMed]
- García, M.L.; Fernández, A.; Solas, M.T. Mitochondria, motor neurons and aging. J. Neurol. Sci. 2013, 330, 18–26. [Google Scholar] [CrossRef]
- Jang, Y.C.; Van Remmen, H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011, 46, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.L.; Rygiel, K.; Purves-Smith, F.M.; Solbak, N.M.; Turnbull, D.M.; Hepple, R.T. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 2012, 7, e29082. [Google Scholar] [CrossRef]
- Clark, D.J.; Fielding, R.A. Neuromuscular contributions to age-related weakness. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 41–47. [Google Scholar] [CrossRef]
- Rosso, A.L.; Studenski, S.A.; Chen, W.G.; Aizenstein, H.J.; Alexander, N.B.; Bennett, D.A.; Black, S.E.; Camicioli, R.; Carlson, M.C.; Ferrucci, L.; et al. Aging, the central nervous system, and mobility. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1379–1386. [Google Scholar] [CrossRef]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Rudnicki, M.A. Satellite cells: The architects of skeletal muscle. Curr. Top. Dev. Biol. 2014, 107, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wehling-Henricks, M.; Welc, S.S.; Fisher, A.L.; Zuo, Q.; Tidball, J.G. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J. 2019, 33, 1415–1427. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Muñoz-Cánoves, P. Regenerative decline of stem cells in sarcopenia. Mol. Asp. Med. 2016, 50, 109–117. [Google Scholar] [CrossRef]
- Hong, X.; Campanario, S.; Ramírez-Pardo, I.; Grima-Terrén, M.; Isern, J.; Muñoz-Cánoves, P. Stem cell aging in the skeletal muscle: The importance of communication. Ageing Res. Rev. 2022, 73, 101528. [Google Scholar] [CrossRef]
- Chiristov, C.; Chrétien, F.; Abou-Khalil, R.; Bassez, G.; Vallet, G.; Authier, F.J.; Bassaglia, Y.; Shinin, V.; Tajbakhsh, S.; Chazaud, B.; et al. Muscle satellite cells and endothelial cells: Close neighbors and privileged partners. Mol. Biol. Cell 2007, 18, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Joe, A.W.B.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M.V. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ziemkiewicz, N.; Hilliard, G.; Pullen, N.A.; Garg, K. The role of innate and adaptive immune cells in skeletal muscle regeneration. Int. J. Mol. Sci. 2021, 22, 3265. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Di Maggio, R.; Benedetti, A.; Morroni, J.; Bouche, M.; Lozanoska-Ochser, B. Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight 2020, 5, e130807. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Verma, M.; Asakura, Y.; Murakonda, B.S.R.; Pengo, T.; Latroche, C.; Chazaud, B.; McLoon, L.K.; Asakura, A. Muscle satellite cell cross-talk with a vascular niche maintains quiescence via VEGF and Notch signaling. Cell Stem Cell 2018, 23, 530–543.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Noguchi, Y.-T.; Nakayama, H.; Kaji, T.; Tsujikawa, K.; Ikemoto-Uezumi, M.; Uezumi, A.; Okada, Y.; Doi, T.; Watanabe, S.; et al. The CalcR-PKA-Yap1 axis is critical for maintaining quiescence in muscle stem cells. Cell Rep. 2019, 29, 2154–2163.e5. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.A.; Zwetsloot, K.A.; Westerkamp, L.M.; Hickner, R.C.; Pofahl, W.E.; Gavin, T.P. Lower skeletal muscle capillarization and VEGF expression in aged vs. young men. J. Appl. Physiol. 2006, 100, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C. Autophagy and stem cells: Self-eating for self-renewal. Front. Cell Dev. Biol. 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Chen, J.; Gu, L.; Dan, X.; Zhang, C.; Yang, Y. New insights into mitophagy and stem cells. Stem Cell Res. Ther. 2021, 12, 452. [Google Scholar] [CrossRef]
- Fiacco, E.; Castagnetti, F.; Bianconi, V.; Madaro, L.; De Bardi, M.; Nazio, F.; D’Amico, A.; Bertini, E.; Cecconi, F.; Puri, P.L.; et al. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. 2016, 23, 1839–1849. [Google Scholar] [CrossRef]
- García-Prat, L.; Muñoz-Cánoves, P.; Martinez-Vicente, M. Dysfunctional autophagy is a driver of muscle stem cell functional decline with aging. Autophagy 2016, 12, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Victor, P.; Gutarra, S.; García-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardí, M.; Ballestar, E.; González, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef]
- Wang, Y.; Wehling-Henricks, M.; Samengo, G.; Tidball, J.G. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 2015, 14, 678–688. [Google Scholar] [CrossRef]
- Tidball, J.G. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017, 17, 165–178. [Google Scholar] [CrossRef]
- Tidball, J. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Voss, J.G.; Tsuji, J.; Fulkerson, N.D.; Soulakova, J.; Schneider, B.S.P. Time course of chemokine expression and leukocyte infiltration after acute skeletal muscle injury in mice. Innate Immun. 2015, 21, 266–274. [Google Scholar] [CrossRef]
- Brigitte, M.; Schilte, C.; Plonquet, A.; Baba-Amer, Y.; Henri, A.; Charlier, C.; Tajbakhsh, S.; Albert, M.; Gherardi, R.K.; Chrétien, F. Muscle resident macrophages control the immune cell reaction in a mouse model of notexin-induced myoinjury. Arthritis Rheum. 2010, 62, 268–279. [Google Scholar] [CrossRef]
- Tidball, J. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R345–R353. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; Van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Bencze, M.; Negroni, E.; Vallese, D.; Yacoub-Youssef, H.; Chaouch, S.; Wolff, A.; Aamiri, A.; Di Santo, J.P.; Chazaud, B.; Butler-Browne, G.; et al. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol. Ther. 2012, 20, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, A.; Corna, G.; Rigamonti, E.; Basso, V.; Vezzoli, M.; Monno, A.; Almada, A.E.; Mondino, A.; Wagers, A.J.; Manfredi, A.A.; et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS ONE 2015, 10, e0128094. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Wehling-Henricks, M.; Villalta, S.A.; Wang, Y.; Tidball, J.G. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol. 2012, 189, 3669–3680. [Google Scholar] [CrossRef]
- Muñoz-Cánoves, P.; Neves, J.; Sousa-Victor, P. Understanding muscle regenerative decline with aging: New approaches to bring back youthfulness to aged stem cells. FEBS J. 2020, 287, 406–416. [Google Scholar] [CrossRef]
- Tidball, J.G.; Flores, I.; Welc, S.S.; Wehling-Henricks, M.; Ochi, E. Aging of the immune system and impaired muscle regeneration: A failure of immunomodulation of adult myogenesis. Exp. Gerontol. 2021, 145, 111200. [Google Scholar] [CrossRef]
- Patsalos, A.; Simandi, Z.; Hays, T.T.; Peloquin, M.; Hajian, M.; Restrepo, I.; Coen, P.M.; Russell, A.J.; Nagy, L. In vivo GDF3 administration abrogates aging related muscle regeneration delay following acute sterile injury. Aging Cell 2018, 17, e12815. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, P.; Pishesha, N.; Wijaya, D.; Conboy, I.M. Age dependent increase in the levels of osteopontin inhibits skeletal muscle regeneration. Aging 2012, 4, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-Y.; Driscoll, R.K.; Piao, Y.; Chia, C.W.; Gorospe, M.; Ferrucci, L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 2019, 18, e13032. [Google Scholar] [CrossRef]
- Wang, H.; Melton, D.W.; Porter, L.; Sarwar, Z.U.; McManus, L.M.; Shireman, P.K. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am. J. Pathol. 2014, 184, 1167–1184. [Google Scholar] [CrossRef]
- Bouchè, M.; Lozanoska-Ochser, B.; Proietti, D.; Madaro, L. Do neurogenic and cancer-induced muscle atrophy follow common or divergent paths? Eur. J. Transl. Myol. 2018, 28, 7931. [Google Scholar] [CrossRef] [PubMed]
- Sloboda, D.D.; Brown, L.A.; Brooks, S. V Myeloid cell responses to contraction-induced injury differ in muscles of young and old mice. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 2011, 187, 4835–4843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cheng, N.; Qiao, B.; Zhang, F.; Wu, J.; Liu, C.; Li, Y.; Du, J. Age-related decline of interferon-gamma responses in macrophage impairs satellite cell proliferation and regeneration. J. Cachexia Sarcopenia Muscle 2020, 11, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, J.R.; Kaluhiokalani, J.P.; Hafen, P.S.; Deyhle, M.R.; Parcell, A.C.; Hyldahl, R.D. An altered response in macrophage phenotype following damage in aged human skeletal muscle: Implications for skeletal muscle repair. FASEB J. 2019, 33, 10353–10368. [Google Scholar] [CrossRef] [PubMed]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef]
- Panduro, M.; Benoist, C.; Mathis, D. Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2585–E2593. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.; Giagnacovo, M.; Costanzo, M.; Fattoretti, P.; Zancanaro, C.; Pellicciari, C.; Malatesta, M. Adapted physical exercise enhances activation and differentiation potential of satellite cells in the skeletal muscle of old mice. J. Anat. 2016, 228, 771–783. [Google Scholar] [CrossRef]
- Joanisse, S.; Nederveen, J.P.; Baker, J.M.; Snijders, T.; Iacono, C.; Parise, G. Exercise conditioning in old mice improves skeletal muscle regeneration. FASEB J. 2016, 30, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Chikenji, T.S.; Matsumura, T.; Nakano, M.; Fujimiya, M. Exercise enhances skeletal muscle regeneration by promoting senescence in fibro-adipogenic progenitors. Nat. Commun. 2020, 11, 889. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, R.; Feng, Y.; Cheng, L. Molecular mechanisms of exercise contributing to tissue regeneration. Signal Transduct. Target. Ther. 2022, 7, 383. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed]
- Morroni, J.; Benedetti, A.; Esposito, L.; De Bardi, M.; Borsellino, G.; Riera, C.S.; Giordani, L.; Bouche, M.; Lozanoska-Ochser, B. Injury-experienced satellite cells retain long-term enhanced regenerative capacity. Stem Cell Res. Ther. 2023, 14, 246. [Google Scholar] [CrossRef]
- Peake, J.M.; Neubauer, O.; Della Gatta, P.A.; Nosaka, K. Muscle damage and inflammation during recovery from exercise. J. Appl. Physiol. 2017, 122, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Kosmac, K.; Peck, B.D.; Walton, R.G.; Mula, J.; Kern, P.A.; Bamman, M.M.; Dennis, R.A.; Jacobs, C.A.; Lattermann, C.; Johnson, D.L.; et al. Immunohistochemical identification of human skeletal muscle macrophages. Bio-Protocol 2018, 8, e2883. [Google Scholar] [CrossRef]
- Walton, R.G.; Kosmac, K.; Mula, J.; Fry, C.S.; Peck, B.D.; Groshong, J.S.; Finlin, B.S.; Zhu, B.; Kern, P.A.; Peterson, C.A. Human skeletal muscle macrophages increase following cycle training and are associated with adaptations that may facilitate growth. Sci. Rep. 2019, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.D.; Murach, K.A.; Walton, R.G.; Simmons, A.J.; Long, D.E.; Kosmac, K.; Dungan, C.M.; Kern, P.A.; Bamman, M.M.; Peterson, C.A. A Muscle cell-macrophage axis involving matrix metalloproteinase 14 facilitates extracellular matrix remodeling with mechanical loading. FASEB J. 2022, 36, e22155. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Monzel, A.S.; Enríquez, J.A.; Picard, M. Multifaceted mitochondria: Moving mitochondrial science beyond function and dysfunction. Nat. Metab. 2023, 5, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: A review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.F.; Barrett, J.N.; David, G. Mitochondria in motor nerve terminals: Function in health and in mutant superoxide dismutase 1 mouse models of familial ALS. J. Bioenerg. Biomembr. 2011, 43, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Boveris, A. Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr. 2009, 41, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R. Motor unit and neuromuscular junction remodeling with aging. Curr. Aging Sci. 2012, 4, 209–220. [Google Scholar] [CrossRef]
- Choi, S.; Reiter, D.A.; Shardell, M.; Simonsick, E.M.; Studenski, S.; Spencer, R.G.; Fishbein, K.W.; Ferrucci, L. 31P magnetic resonance spectroscopy assessment of muscle bioenergetics as a predictor of gait speed in the Baltimore longitudinal study of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1638–1645. [Google Scholar] [CrossRef]
- Tian, Q.; Mitchell, B.A.; Zampino, M.; Fishbein, K.W.; Spencer, R.G.; Ferrucci, L. Muscle mitochondrial energetics predicts mobility decline in well-functioning older adults: The Baltimore Longitudinal Study of Aging. Aging Cell 2022, 21, e13552. [Google Scholar] [CrossRef] [PubMed]
- Zane, A.C.; Reiter, D.A.; Shardell, M.; Cameron, D.; Simonsick, E.M.; Fishbein, K.W.; Studenski, S.A.; Spencer, R.G.; Ferrucci, L. Muscle strength mediates the relationship between mitochondrial energetics and walking performance. Aging Cell 2017, 16, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D’Amico, D. Mitophagy in human health, ageing and disease. Nat. Metab. 2023, 5, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.M.; Williams, S.L.; Hida, A.; Peacock, J.D.; Prolla, T.A.; Lincoln, J.; Moraes, C.T. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum. Mol. Genet. 2012, 21, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.-D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Wang, J.; Zhang, D.; Wu, H.; Li, W.; Wei, H.; Ta, N.; Fan, Y.; Liu, Y.; et al. Mitophagy receptor FUNDC1 is regulated by PGC-1α/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021, 22, e50629. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Leeuwenburgh, C.; Coelho-Junior, H.J.; Bernabei, R.; Landi, F.; Marzetti, E. Targeting mitochondrial quality control for treating sarcopenia: Lessons from physical exercise. Expert Opin. Ther. Targets 2019, 23, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Shi, Z.; Liu, X.; Sun, C. The research progress of mitochondrial transplantation in the treatment of mitochondrial defective diseases. Int. J. Mol. Sci. 2024, 25, 1175. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef]
- D’Amato, M.; Morra, F.; Di Meo, I.; Tiranti, V. Mitochondrial transplantation in mitochondrial medicine: Current challenges and future perspectives. Int. J. Mol. Sci. 2023, 24, 1969. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Slavin, M.B.; Hood, D.A. Muscle mitochondrial transplantation can rescue and maintain cellular homeostasis. Am. J. Physiol. Cell Physiol. 2023, 325, C862–C884. [Google Scholar] [CrossRef] [PubMed]
- Orfany, A.; Arriola, C.G.; Doulamis, I.P.; Guariento, A.; Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Rogers, C.; Del Nido, P.J.; et al. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 2020, 71, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Boutonnet, L.; Mallard, J.; Charles, A.-L.; Hucteau, E.; Gény, B.; Lejay, A.; Grandperrin, A. Autologous mitochondrial transplantation in male mice as a strategy to prevent deleterious effects of peripheral ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2024, 326, C449–C456. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Endo, Y.; Nakamura, E.; Kuschner, C.E.; Kazmi, J.; Singh, P.; Yin, T.; Becker, L.B.; Hayashida, K. Therapeutic potential of mitochondrial transplantation in modulating immune responses post-cardiac arrest: A narrative review. J. Transl. Med. 2024, 22, 230. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, J.M.; Min, K.; Choi, Y.-S. Xenogeneic Transplantation of Mitochondria Induces Muscle Regeneration in an In Vivo Rat Model of Dexamethasone-Induced Atrophy. J. Muscle Res. Cell Motil. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, J.; Ni, H.; Zhang, L.; Wang, J.; Li, Y.; Jiang, W.; Wu, Z.; Zhou, M. Mitochondrial transplantation reduces lower limb ischemia-reperfusion injury by increasing skeletal muscle energy and adipocyte browning. Mol. Ther. Methods Clin. Dev. 2023, 31, 101152. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzetti, E.; Lozanoska-Ochser, B.; Calvani, R.; Landi, F.; Coelho-Júnior, H.J.; Picca, A. Restoring Mitochondrial Function and Muscle Satellite Cell Signaling: Remedies against Age-Related Sarcopenia. Biomolecules 2024, 14, 415. https://doi.org/10.3390/biom14040415
Marzetti E, Lozanoska-Ochser B, Calvani R, Landi F, Coelho-Júnior HJ, Picca A. Restoring Mitochondrial Function and Muscle Satellite Cell Signaling: Remedies against Age-Related Sarcopenia. Biomolecules. 2024; 14(4):415. https://doi.org/10.3390/biom14040415
Chicago/Turabian StyleMarzetti, Emanuele, Biliana Lozanoska-Ochser, Riccardo Calvani, Francesco Landi, Hélio José Coelho-Júnior, and Anna Picca. 2024. "Restoring Mitochondrial Function and Muscle Satellite Cell Signaling: Remedies against Age-Related Sarcopenia" Biomolecules 14, no. 4: 415. https://doi.org/10.3390/biom14040415