
Changes in Internal Structure and Dynamics upon Binding Stabilise the Nematode Anticoagulant NAPc2


Abstract
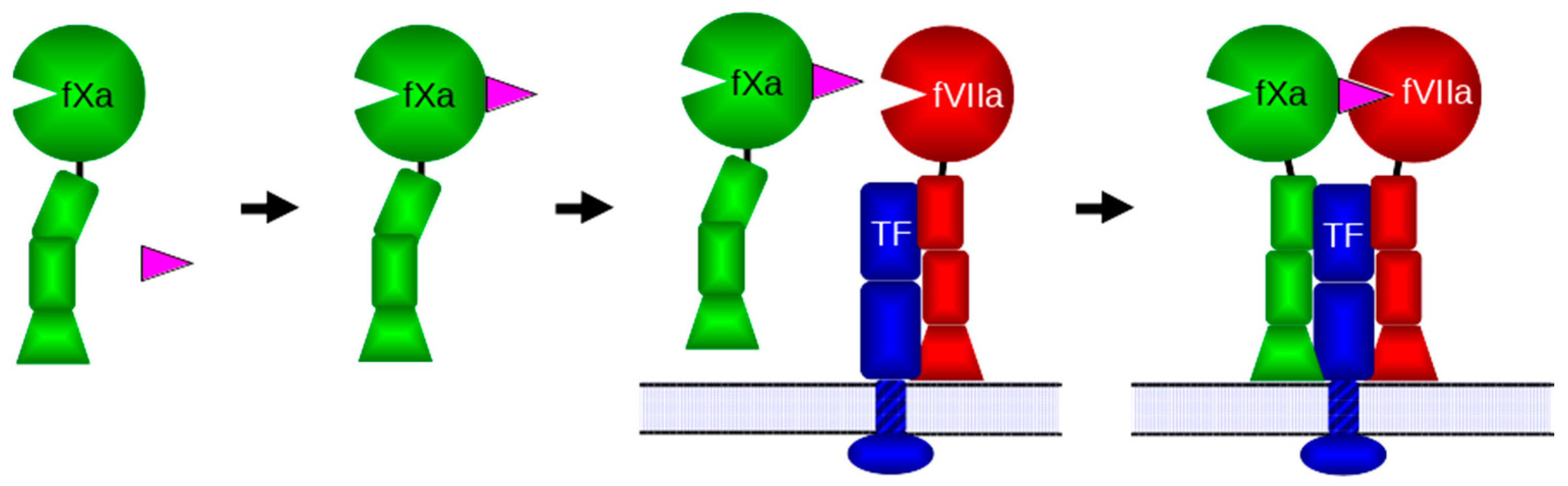
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.2. Plasmid Construction
2.3. Protein Expression and Purification
2.4. Prothrombin Time Coagulation Assays
3. Results
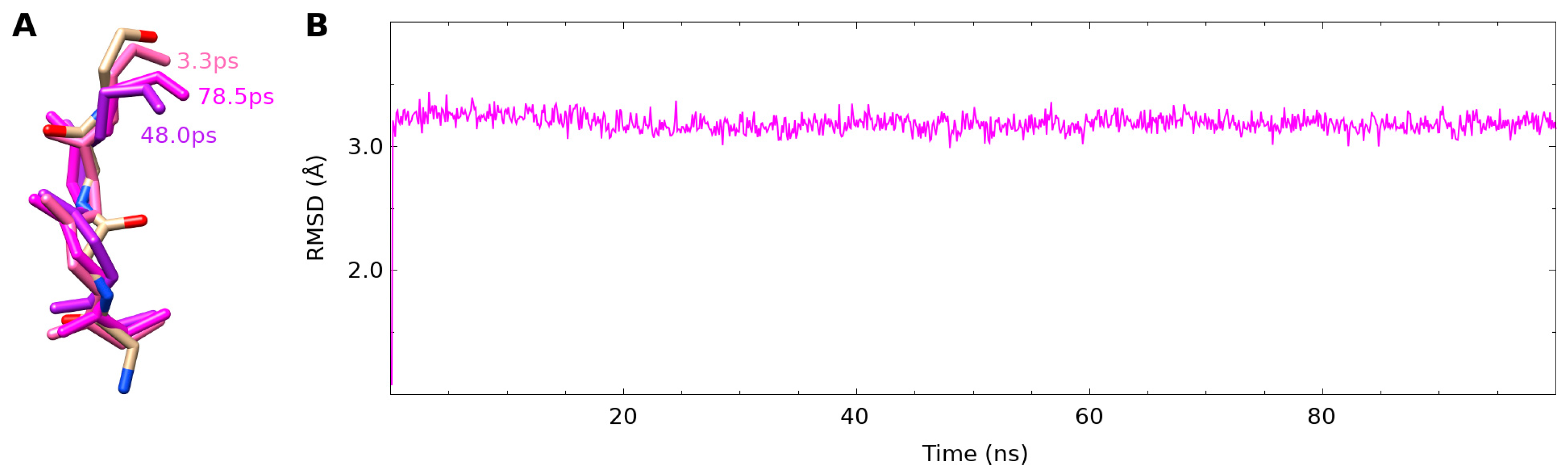
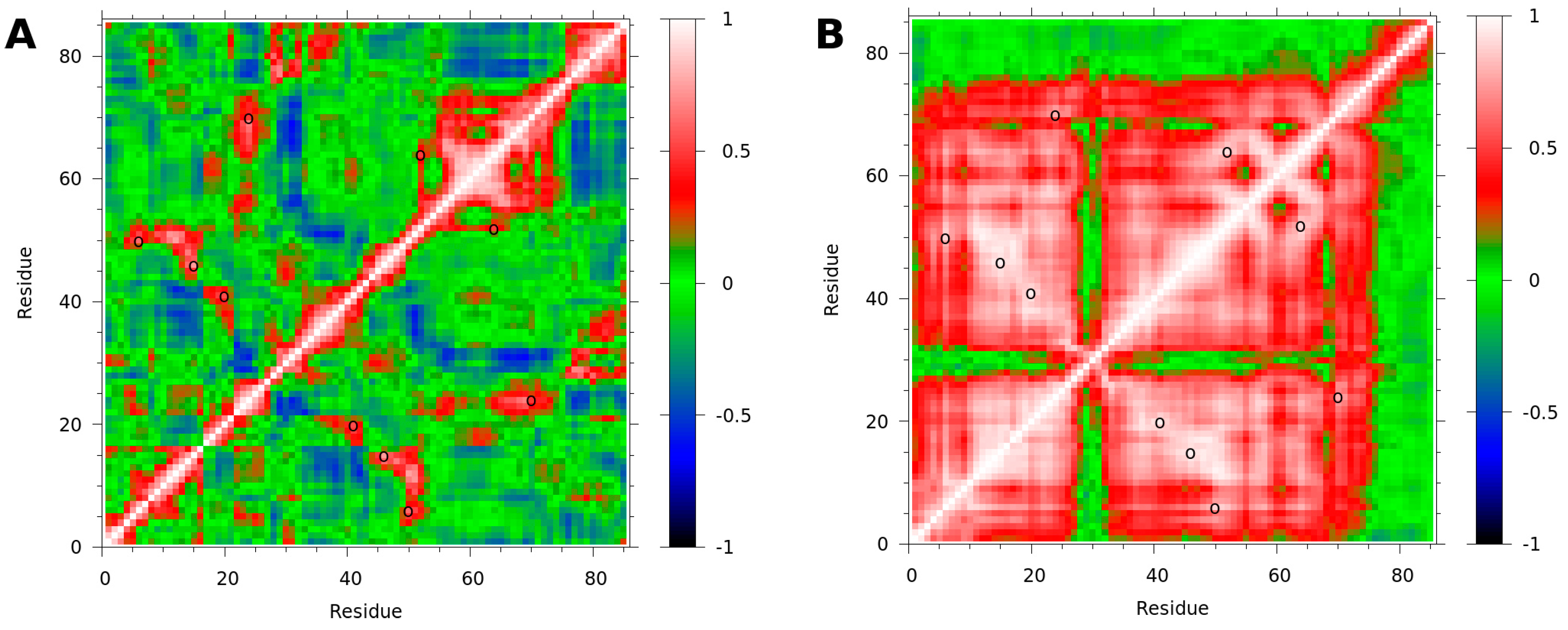
3.1. Simulations Capture Mobility of NAPc2
3.2. The fXa Bound Conformation Was Not Present in the Free NAPC2 Simulation
3.3. Binding to fXa Stabilizes NAPc2
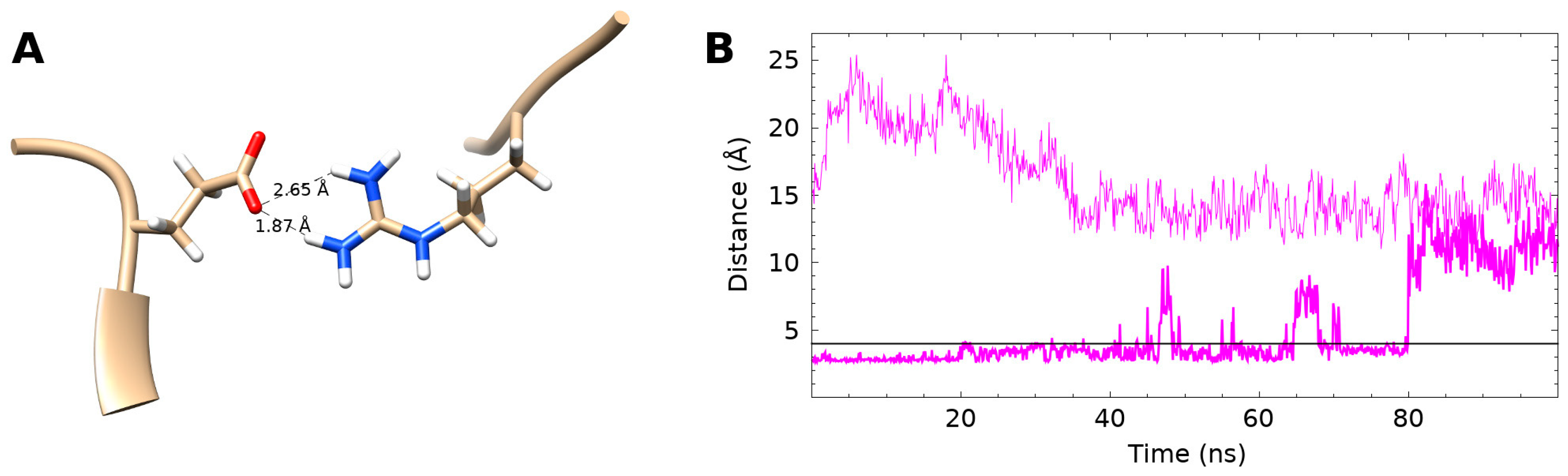
3.4. Internal Salt Bridge Found in Bound Conformation
3.5. Internal Salt Bridge Required for Maximal Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rathbun, S. The Surgeon General’s Call to Action to Prevent Deep Vein Thrombosis and Pulmonary Embolism. Circulation 2009, 119, e480–e482. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, J.; O’Donnell, M.; Eikelboom, J.W. Beyond Unfractionated Heparin and Warfarin: Current and Future Advances. Circulation 2007, 116, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Stassens, P.; Bergum, P.W.; Gansemans, Y.; Jespers, L.; Laroche, Y.; Huang, S.; Maki, S.; Messens, J.; Lauwereys, M.; Cappello, M.; et al. Anticoagulant repertoire of the hookworm Ancylostoma caninum. Proc. Natl. Acad. Sci. USA 1996, 93, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.Y.; Vlasuk, G.P. Recombinant nematode anticoagulant protein c2 and other inhibitors targeting blood coagulation factor VIIa/tissue factor. J. Intern. Med. 2003, 254, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhan, B.; Mayor, R.S.; Gillespie, P.; Keegan, B.; Bottazzi, M.E.; Hotez, P. Ac-AP-12, a novel factor Xa anticoagulant peptide from the esophageal glands of adult Ancylostoma caninum. Mol. Biochem. Parasitol. 2011, 177, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.T.; Rios-Steiner, J.; Weaver, S.E.; Tulinsky, A.; Geiger, J.H.; Arni, R.K. Intermolecular Interactions and Characterization of the Novel Factor Xa Exosite Involved in Macromolecular Recognition and Inhibition: Crystal Structure of Human Gla-domainless Factor Xa Complexed with the Anticoagulant Protein NAPc2 from the Hematophagous Nematode Ancylostoma caninum. J. Mol. Biol. 2007, 366, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Duggan, B.M.; Dyson, H.J.; Wright, P.E. Inherent flexibility in a potent inhibitor of blood coagulation, recombinant nematode anticoagulant protein c2. Eur. J. Biochem. 1999, 265, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Machado, M.R.; Pantano, S. Split the Charge Difference in Two! A Rule of Thumb for Adding Proper Amounts of Ions in MD Simulations. J. Chem. Theory Comput. 2020, 16, 1367–1372. [Google Scholar] [CrossRef]
- Adelman, S.A.; Doll, J.D. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375–2388. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.C.; Daggett, V. Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles. J. Phys. Chem. B 2018, 122, 6673–6689. [Google Scholar] [CrossRef] [PubMed]
- Habeck, M.; Rieping, W.; Nilges, M. Bayesian estimation of Karplus parameters and torsion angles from three-bond scalar couplings constants. J. Magn. Reson. 2005, 177, 160–165. [Google Scholar] [CrossRef]
- Hünenberger, P.H.; Mark, A.E.; Van Gunsteren, W.F. Fluctuation and Cross-correlation Analysis of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. J. Mol. Biol. 1995, 252, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Ban, X.; Lahiri, P.; Dhoble, A.S.; Li, D.; Gu, Z.; Li, C.; Cheng, L.; Hong, Y.; Li, Z.; Kaustubh, B. Evolutionary Stability of Salt Bridges Hints Its Contribution to Stability of Proteins. Comput. Struct. Biotechnol. J. 2019, 17, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Briggs, J.M. Molecular dynamics simulations of Factor Xa: Insight into conformational transition of its binding subsites. Biopolymers 2008, 89, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Strynadka, N.C.; Bernard, V.D.; Peanasky, R.J.; James, M.N. The molecular structure of the complex of Ascaris chymotrypsin/elastase inhibitor with porcine elastase. Structure 1994, 2, 679–689. [Google Scholar] [CrossRef]
- Rios-Steiner, J.L.; Murakami, M.T.; Tulinsky, A.; Arni, R.K. Active and Exo-site Inhibition of Human Factor Xa: Structure of des-Gla Factor Xa Inhibited by NAP5, a Potent Nematode Anticoagulant Protein from Ancylostoma caninum. J. Mol. Biol. 2007, 371, 774–786. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Clore, G.M.; Gronenborn, A.M. High-resolution structure of Ascaris trypsin inhibitor in solution: Direct evidence for a pH-induced conformational transition in the reactive site. Structure 1994, 2, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Cierpicki, T.; Bania, J.; Otlewski, J. NMR solution structure of Apis mellifera chymotrypsin/cathepsin G inhibitor-1 (AMCI-1): Structural similarity with Ascaris protease inhibitors. Protein Sci. 2000, 9, 976–984. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Daly, N.L.; Scanlon, M.J.; Craik, D.J. Solution Structure of BSTI: A New Trypsin Inhibitor from Skin Secretions of Bombina bombina. Biochemistry 2001, 40, 4601–4609. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAPc2 | fXa | NAPc2-fXa | |
|---|---|---|---|
| PDB code | 1COU 1 | 2H9E | 2H9E |
| PDB method | NMR | X-ray | X-ray |
| residues | 85 | 281 | 366 |
| protein atoms | 1267 | 4312 | 5579 |
| counterions | 18 Na+, 6 Cl− | 28 Na+, 29 Cl− | 63 Na+, 50 Cl− |
| water molecules | 4481 | 10,434 | 15,885 |
| box size (Å) | 33 × 28 × 44 | 47 × 65 × 53 | 53 × 57 × 80 |
| simulation length (ns) | 100 | 100 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodward, E.; Duggan, B.M. Changes in Internal Structure and Dynamics upon Binding Stabilise the Nematode Anticoagulant NAPc2. Biomolecules 2024, 14, 421. https://doi.org/10.3390/biom14040421
Woodward E, Duggan BM. Changes in Internal Structure and Dynamics upon Binding Stabilise the Nematode Anticoagulant NAPc2. Biomolecules. 2024; 14(4):421. https://doi.org/10.3390/biom14040421
Chicago/Turabian StyleWoodward, Elaine, and Brendan M. Duggan. 2024. "Changes in Internal Structure and Dynamics upon Binding Stabilise the Nematode Anticoagulant NAPc2" Biomolecules 14, no. 4: 421. https://doi.org/10.3390/biom14040421
APA StyleWoodward, E., & Duggan, B. M. (2024). Changes in Internal Structure and Dynamics upon Binding Stabilise the Nematode Anticoagulant NAPc2. Biomolecules, 14(4), 421. https://doi.org/10.3390/biom14040421