

Computational Modeling and Characterization of Peptides Derived from Nanobody Complementary-Determining Region 2 (CDR2) Targeting Active-State Conformation of the β2-Adrenergic Receptor (β2AR)

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Informational Spectrum Method (ISM) and Computational Peptide Scanning
2.3. Datasets and Computational Peptide Scanning
2.4. Receptor Preparation
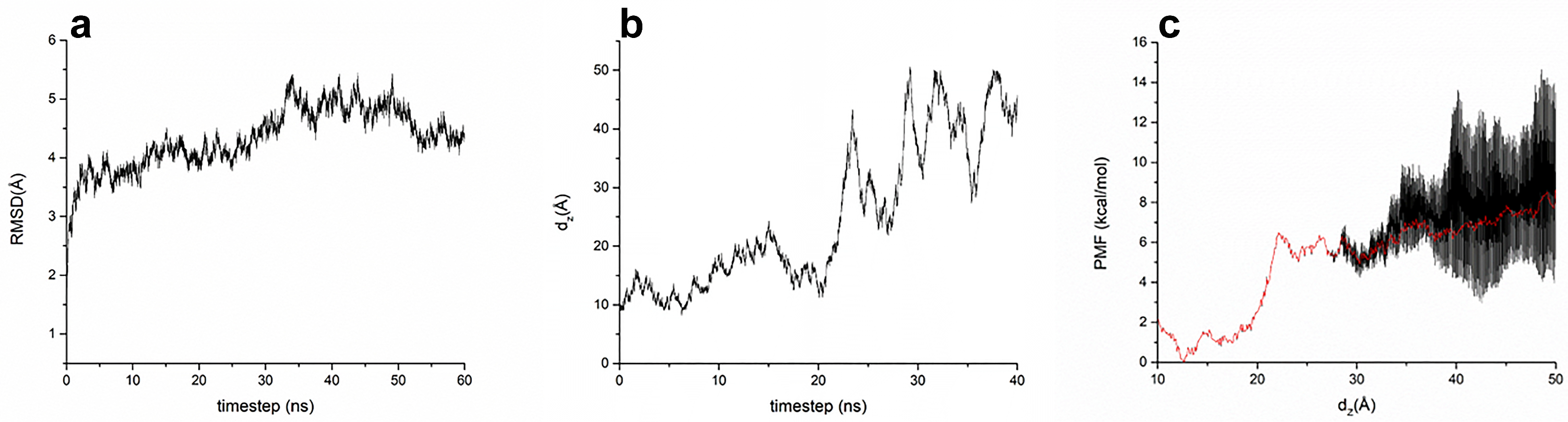
2.5. Molecular Dynamics Simulations
2.6. Fusion Constructs, Cell Culture, and Transfection
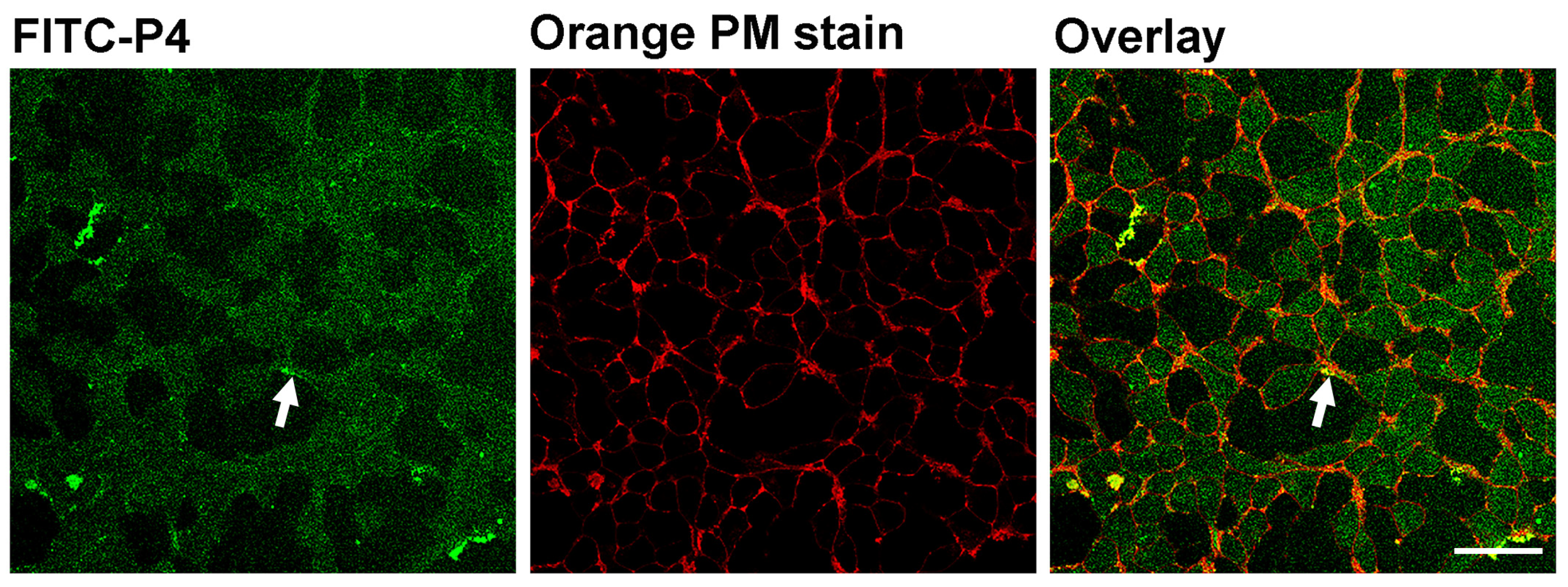
2.7. Confocal Microscopy
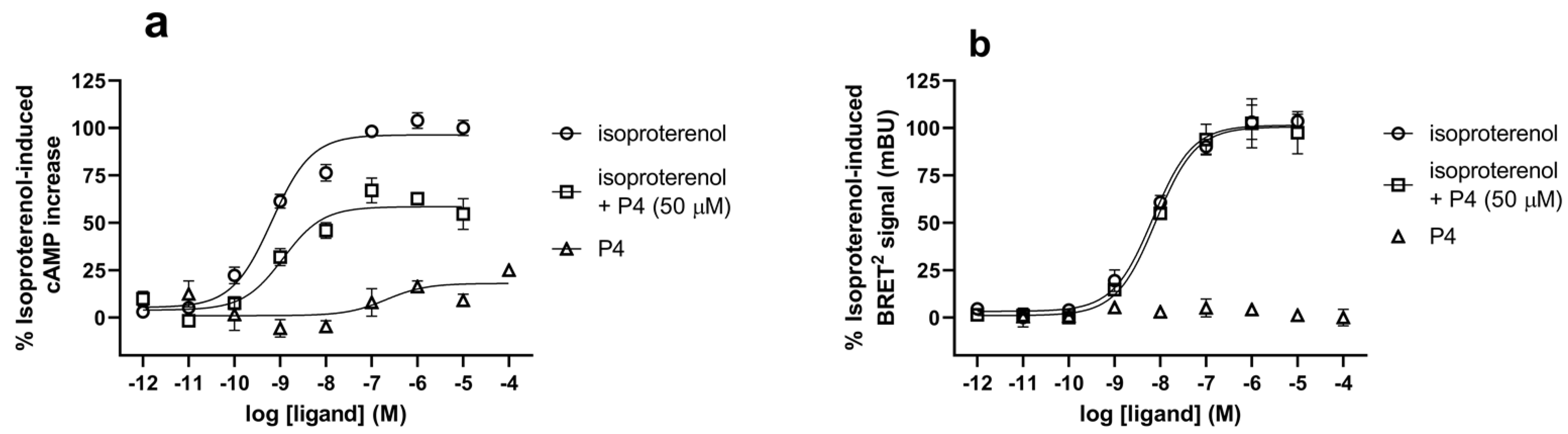
2.8. cAMP Assay
2.9. BRET2-Based β-Arrestin 2 Recruitment Assay
2.10. Microscale Thermophoresis (MST)
3. Results
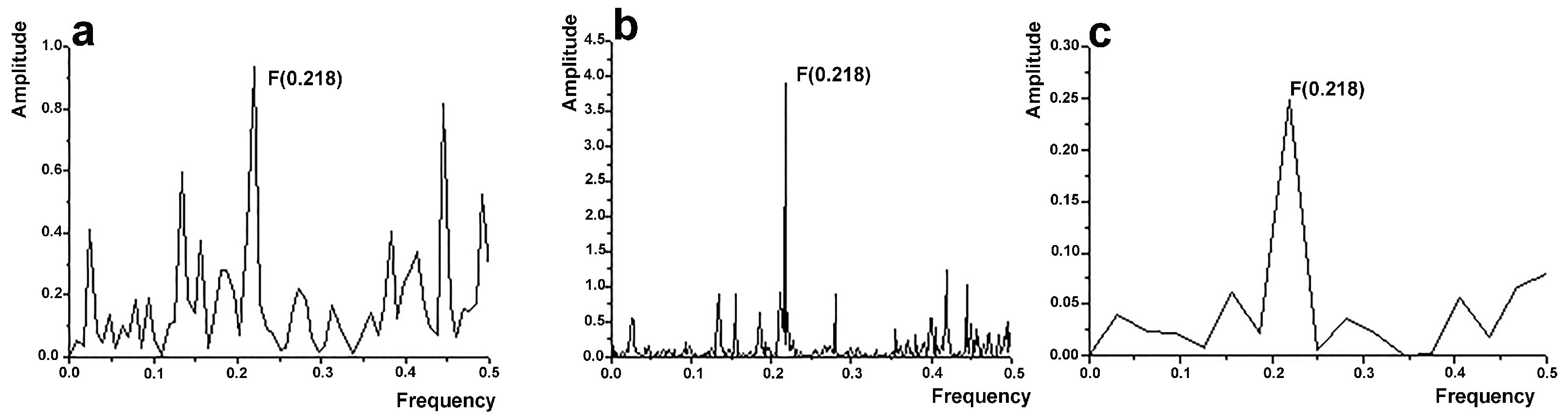
3.1. Computational Design and Modeling
3.2. In Vitro Characterization of P4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassanzadeh-Ghassabeh, G.; Devoogdt, N.; De Pauw, P.; Vincke, C.; Muyldermans, S. Nanobodies and their potential applications. Nanomedicine 2013, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.T.T.; Huynh, V.Q.; Vo, N.T.; Nguyen, H.D. Studying the characteristics of nanobody CDR regions based on sequence analysis in combination with 3D structures. J. Genet. Eng. Biotechnol. 2022, 20, 157. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.L.; Davis, M.M. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 2000, 13, 37–45. [Google Scholar] [CrossRef]
- Chen, C.; Roberts, V.A.; Rittenberg, M.B. Generation and analysis of random point mutations in an antibody CDR2 sequence: Many mutated antibodies lose their ability to bind antigen. J. Exp. Med. 1992, 176, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.J.; Vance, D.J.; Kelow, S.; Angalakurthi, S.K.; Nguyen, S.; Davis, S.A.; Rong, Y.; Middaugh, C.R.; Weis, D.D.; Dunbrack, R., Jr.; et al. Contribution of an unusual CDR2 element of a single domain antibody in ricin toxin binding affinity and neutralizing activity. Protein Eng. Des. Sel. PEDS 2018, 31, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef]
- Heukers, R.; De Groof, T.W.M.; Smit, M.J. Nanobodies detecting and modulating GPCRs outside in and inside out. Curr. Opin. Cell Biol. 2019, 57, 115–122. [Google Scholar] [CrossRef]
- Cromie, K.D.; Van Heeke, G.; Boutton, C. Nanobodies and their Use in GPCR Drug Discovery. Curr. Top. Med. Chem. 2015, 15, 2543–2557. [Google Scholar] [CrossRef]
- Mujić-Delić, A.; de Wit, R.H.; Verkaar, F.; Smit, M.J. GPCR-targeting nanobodies: Attractive research tools, diagnostics, and therapeutics. Trends Pharmacol. Sci. 2014, 35, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kobilka, B.K.; Steyaert, J. Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Feld, A.P. Nanobodies as Probes and Modulators of Cardiovascular G Protein-Coupled Receptors. J. Cardiovasc. Pharmacol. 2022, 80, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Wingler, L.M.; Strachan, R.T.; Rasmussen, S.G.; Pardon, E.; Ahn, S.; Steyaert, J.; Kobilka, B.K.; Lefkowitz, R.J. Regulation of beta2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol. Pharmacol. 2014, 85, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Van Holsbeeck, K.; Martins, J.C.; Ballet, S. Downsizing antibodies: Towards complementarity-determining region (CDR)-based peptide mimetics. Bioorganic Chem. 2022, 119, 105563. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Moors, S.L.C.; Danielsen, M.; Betti, C.; Fabris, C.; Sejer Pedersen, D.; Pardon, E.; Peyressatre, M.; Feher, K.; Martins, J.C.; et al. Rational Design of Nanobody80 Loop Peptidomimetics: Towards Biased beta2 Adrenergic Receptor Ligands. Chem. A Eur. J. 2017, 23, 9632–9640. [Google Scholar] [CrossRef] [PubMed]
- Sencanski, M.; Glisic, S.; Šnajder, M.; Veljkovic, N.; Poklar Ulrih, N.; Mavri, J.; Vrecl, M. Computational design and characterization of nanobody-derived peptides that stabilize the active conformation of the β(2)-adrenergic receptor (β(2)-AR). Sci. Rep. 2019, 9, 16555. [Google Scholar] [CrossRef]
- Karami, E.; Sabatier, J.M.; Behdani, M.; Irani, S.; Kazemi-Lomedasht, F. A nanobody-derived mimotope against VEGF inhibits cancer angiogenesis. J. Enzym. Inhib. Med. Chem. 2020, 35, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Schmier, S.; Mostafa, A.; Haarmann, T.; Bannert, N.; Ziebuhr, J.; Veljkovic, V.; Dietrich, U.; Pleschka, S. In Silico Prediction and Experimental Confirmation of HA Residues Conferring Enhanced Human Receptor Specificity of H5N1 Influenza A Viruses. Sci. Rep. 2015, 5, 11434. [Google Scholar] [CrossRef]
- Veljkovic, N.; Glisic, S.; Prljic, J.; Perovic, V.; Botta, M.; Veljkovic, V. Discovery of new therapeutic targets by the informational spectrum method. Curr. Protein Pept. Sci. 2008, 9, 493–506. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, N.; Glisic, S.; Perovic, V.; Veljkovic, V. The role of long-range intermolecular interactions in discovery of new drugs. Expert Opin. Drug Discov. 2011, 6, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Doliana, R.; Veljkovic, V.; Prljic, J.; Veljkovic, N.; De Lorenzo, E.; Mongiat, M.; Ligresti, G.; Marastoni, S.; Colombatti, A. EMILINs interact with anthrax protective antigen and inhibit toxin action in vitro. Matrix Biol. J. Int. Soc. Matrix Biol. 2008, 27, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Blagotinšek Cokan, K.; Mavri, M.; Rutland, C.S.; Glišić, S.; Senćanski, M.; Vrecl, M.; Kubale, V. Critical Impact of Different Conserved Endoplasmic Retention Motifs and Dopamine Receptor Interacting Proteins (DRIPs) on Intracellular Localization and Trafficking of the D(2) Dopamine Receptor (D(2)-R) Isoforms. Biomolecules 2020, 10, 1355. [Google Scholar] [CrossRef] [PubMed]
- Mavri, M.; Kubale, V.; Depledge, D.P.; Zuo, J.; Huang, C.A.; Breuer, J.; Vrecl, M.; Jarvis, M.A.; Jovičić, E.J.; Petan, T.; et al. Epstein-Barr Virus-Encoded BILF1 Orthologues from Porcine Lymphotropic Herpesviruses Display Common Molecular Functionality. Front. Endocrinol. 2022, 13, 862940. [Google Scholar] [CrossRef] [PubMed]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Mandic, M.; Drinovec, L.; Glisic, S.; Veljkovic, N.; Nohr, J.; Vrecl, M. Demonstration of a direct interaction between beta2-adrenergic receptor and insulin receptor by BRET and bioinformatics. PLoS ONE 2014, 9, e112664. [Google Scholar] [CrossRef]
- Vrecl, M.; Drinovec, L.; Elling, C.; Heding, A. Opsin oligomerization in a heterologous cell system. J. Recept. Signal Transduct. Res. 2006, 26, 505–526. [Google Scholar] [CrossRef] [PubMed]
- Vrecl, M.; Jorgensen, R.; Pogacnik, A.; Heding, A. Development of a BRET2 screening assay using beta-arrestin 2 mutants. J. Biomol. Screen. 2004, 9, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Kubale, V.; Blagotinsek, K.; Nohr, J.; Eidne, K.A.; Vrecl, M. The Conserved Arginine Cluster in the Insert of the Third Cytoplasmic Loop of the Long Form of the D(2) Dopamine Receptor (D2L-R) Acts as an Intracellular Retention Signal. Int. J. Mol. Sci. 2016, 17, 1152. [Google Scholar] [CrossRef] [PubMed]
- Vrecl, M.; Norregaard, P.K.; Almholt, D.L.; Elster, L.; Pogacnik, A.; Heding, A. Beta-arrestin-based Bret2 screening assay for the “non”-beta-arrestin binding CB1 receptor. J. Biomol. Screen. 2009, 14, 371–380. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Prah, A.; Purg, M.; Stare, J.; Vianello, R.; Mavri, J. How Monoamine Oxidase A Decomposes Serotonin: An Empirical Valence Bond Simulation of the Reactive Step. J. Phys. Chem. B 2020, 124, 8259–8265. [Google Scholar] [CrossRef] [PubMed]
- Vögele, M.; Zhang, B.W.; Kaindl, J.; Wang, L. Is the Functional Response of a Receptor Determined by the Thermodynamics of Ligand Binding? J. Chem. Theory Comput. 2023, 19, 8414–8422. [Google Scholar] [CrossRef] [PubMed]
- Farooq, Z.; Howell, L.A.; McCormick, P.J. Probing GPCR Dimerization Using Peptides. Front. Endocrinol. 2022, 13, 843770. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Elster, L.; Elling, C.; Heding, A. Bioluminescence resonance energy transfer as a screening assay: Focus on partial and inverse agonism. J. Biomol. Screen. 2007, 12, 41–49. [Google Scholar] [CrossRef]
- Ippolito, M.; De Pascali, F.; Hopfinger, N.; Komolov, K.E.; Laurinavichyute, D.; Reddy, P.A.N.; Sakkal, L.A.; Rajkowski, K.Z.; Nayak, A.P.; Lee, J.; et al. Identification of a β-arrestin-biased negative allosteric modulator for the β(2)-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2023, 120, e2302668120. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, X.; Hilger, D.; Aschauer, P.; Tiemann, J.K.S.; Du, Y.; Liu, H.; Hirata, K.; Sun, X.; Guixà-González, R.; et al. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 2019, 177, 1243–1251.e12. [Google Scholar] [CrossRef] [PubMed]
- Mannes, M.; Martin, C.; Triest, S.; Pia Dimmito, M.; Mollica, A.; Laeremans, T.; Menet, C.J.; Ballet, S. Development of Generic G Protein Peptidomimetics Able to Stabilize Active State G(s) Protein-Coupled Receptors for Application in Drug Discovery. Angew. Chem. Int. Ed. Engl. 2021, 60, 10247–10254. [Google Scholar] [CrossRef]
- Carpenter, B.; Tate, C.G. Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr. Opin. Struct. Biol. 2017, 45, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J., 3rd; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Barak, L.S.; Caron, M.G. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J. Biol. Chem. 2001, 276, 19452–19460. [Google Scholar] [CrossRef]
- Shiina, T.; Kawasaki, A.; Nagao, T.; Kurose, H. Interaction with beta-arrestin determines the difference in internalization behavor between beta1- and beta2-adrenergic receptors. J. Biol. Chem. 2000, 275, 29082–29090. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sencanski, M.; Glisic, S.; Kubale, V.; Cotman, M.; Mavri, J.; Vrecl, M. Computational Modeling and Characterization of Peptides Derived from Nanobody Complementary-Determining Region 2 (CDR2) Targeting Active-State Conformation of the β2-Adrenergic Receptor (β2AR). Biomolecules 2024, 14, 423. https://doi.org/10.3390/biom14040423
Sencanski M, Glisic S, Kubale V, Cotman M, Mavri J, Vrecl M. Computational Modeling and Characterization of Peptides Derived from Nanobody Complementary-Determining Region 2 (CDR2) Targeting Active-State Conformation of the β2-Adrenergic Receptor (β2AR). Biomolecules. 2024; 14(4):423. https://doi.org/10.3390/biom14040423
Chicago/Turabian StyleSencanski, Milan, Sanja Glisic, Valentina Kubale, Marko Cotman, Janez Mavri, and Milka Vrecl. 2024. "Computational Modeling and Characterization of Peptides Derived from Nanobody Complementary-Determining Region 2 (CDR2) Targeting Active-State Conformation of the β2-Adrenergic Receptor (β2AR)" Biomolecules 14, no. 4: 423. https://doi.org/10.3390/biom14040423
APA StyleSencanski, M., Glisic, S., Kubale, V., Cotman, M., Mavri, J., & Vrecl, M. (2024). Computational Modeling and Characterization of Peptides Derived from Nanobody Complementary-Determining Region 2 (CDR2) Targeting Active-State Conformation of the β2-Adrenergic Receptor (β2AR). Biomolecules, 14(4), 423. https://doi.org/10.3390/biom14040423