
The Tumor Microenvironment: Signal Transduction
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Dysregulated Signaling Pathways in Tumor Cells
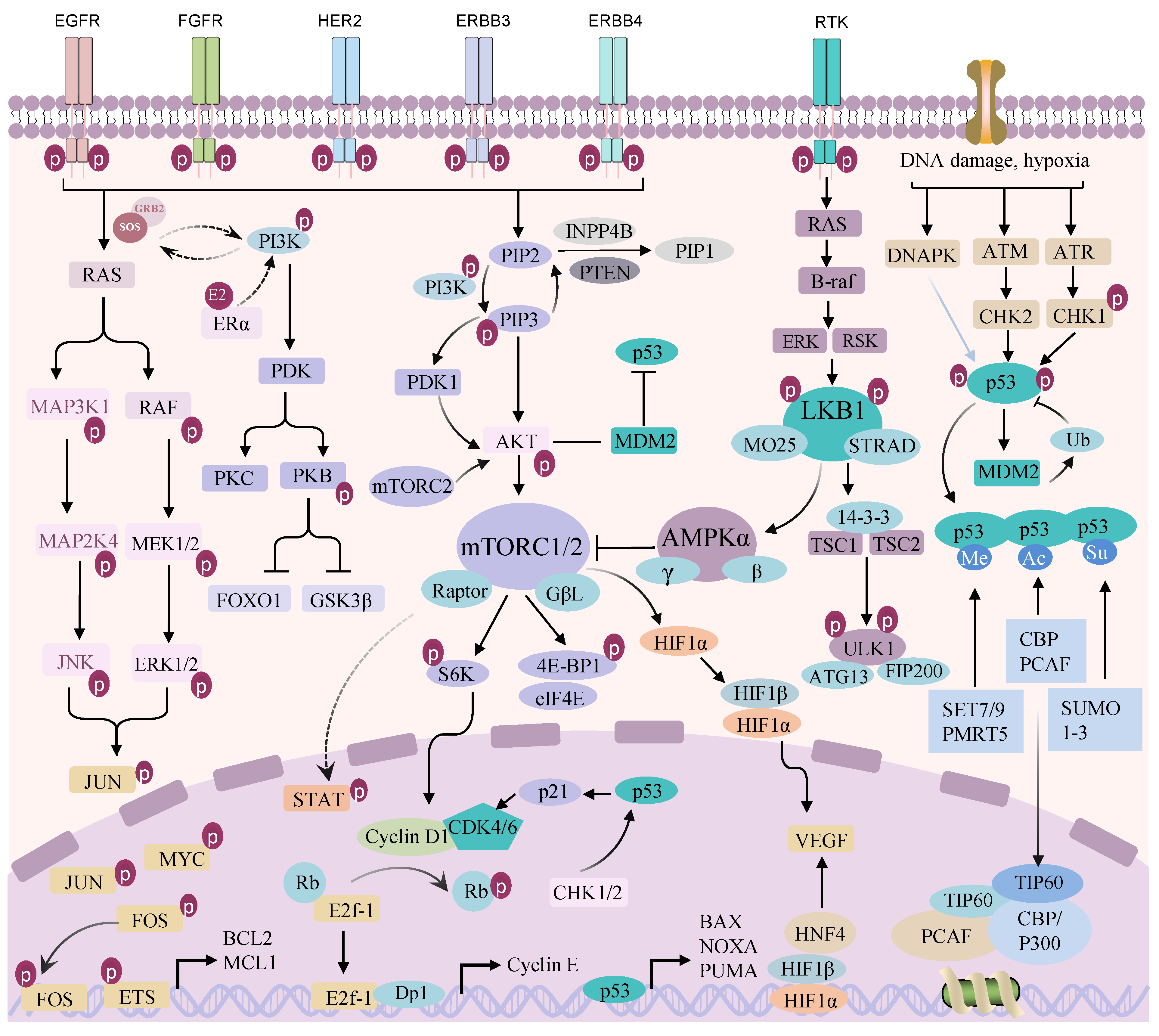
2.1. Epidermal Growth Factor Receptor (ErbB/EGFR) Signaling Pathway
2.2. PI3K/AKT/Mechanistic Target of Rapamycin (mTOR) Signaling Pathway
2.3. RAF/Mitogen-Activated Extracellular Signal-Regulated Kinase (MEK)/ERK and MAPK Signal Pathways
2.4. p53 Signaling Pathway
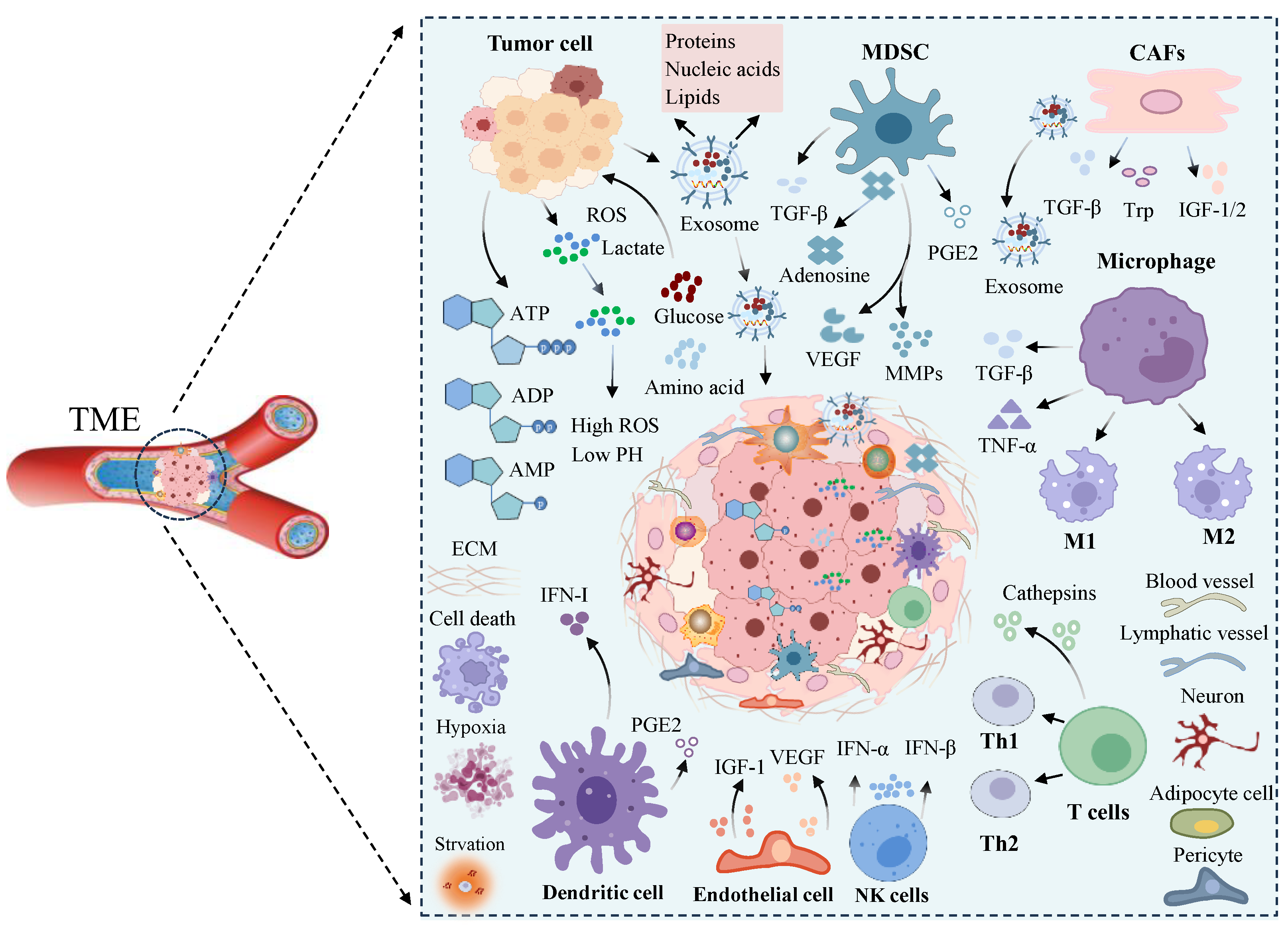
3. Formation and Factors of the Tumor Microenvironment
4. Signaling in the Tumor Microenvironment
4.1. Metabolite-Mediated Signaling
4.2. Signal Transduction Mediated by Soluble Factors
4.3. Cellular Contact-Mediated Molecular Signaling
4.4. Transduction of ER Stress Signals
4.5. Transduction of Notch Signaling
5. Effects of Signaling within the TME on Immune Cells
5.1. T Cells
5.2. Macrophages
5.3. Stromal Cells
6. Summary and Prospect
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Low, V.; Li, Z.; Blenis, J. Metabolite activation of tumorigenic signaling pathways in the tumor microenvironment. Sci. Signal. 2022, 15, eabj4220. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Martin, G.S. Cell signaling and cancer. Cancer Cell 2003, 4, 167–174. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Pitt, J.M.; Marabelle, A.; Eggermont, A.; Soria, J.C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nature Reviews. Cancer 2009, 9, 463–475. [Google Scholar] [PubMed]
- Schlessinger, J.; Ullrich, A. Growth factor signaling by receptor tyrosine kinases. Neuron 1992, 9, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. ErbB Receptors and Cancer. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2017; p. 1652. [Google Scholar]
- Lafky, J.M.; Wilken, J.A.; Baron, A.T.; Maihle, N.J. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim. Biophys. Acta 2008, 1785, 232–265. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.Y.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y.; et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature Reviews. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-L.; He, L.-Y.; Liang, L.; Liu, S.-Y.; Luo, J.; Lv, M.-L.; Cai, Z.-W. Ambra1 regulates apoptosis and chemosensitivity in breast cancer cells through the Akt-FoxO1-Bim pathway. Apoptosis Int. J. Program. Cell Death 2022, 27, 329–341. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Chen, L.; Yang, L.; Yao, L.; Kuang, X.-Y.; Zuo, W.-J.; Li, S.; Qiao, F.; Liu, Y.-R.; Cao, Z.-G.; Zhou, S.-L.; et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 2018, 9, 1357. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.-L.; Huang, K.-H.; Lan, Y.-T.; Lin, C.-H.; Chang, S.-C.; Chen, M.-H.; Chao, Y.; Lin, W.-C.; Lo, S.-S.; Li, A.F.-Y.; et al. Mutations in PI3K/AKT pathway genes and amplifications of PIK3CA are associated with patterns of recurrence in gastric cancers. Oncotarget 2016, 7, 6201–6220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nature Reviews. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wu, G. Targeting mTOR for Anti-Aging and Anti-Cancer Therapy. Molecules 2023, 28, 3157. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Rapa, I.; Saggiorato, E.; Giachino, D.; Palestini, N.; Orlandi, F.; Papotti, M.; Volante, M. Mammalian target of rapamycin pathway activation is associated to RET mutation status in medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2011, 96, 2146–2153. [Google Scholar] [CrossRef]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef]
- Ahn, N.G. The MAP kinase cascade. Discovery of a new signal transduction pathway. Mol. Cell. Biochem. 1993, 127–128, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Leevers, S.J.; Gómez, N.; Nakielny, S.; Cohen, P.; Marshall, C.J. Activation of the MAP kinase pathway by the protein kinase raf. Cell 1992, 71, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Yap, J.L.; Worlikar, S.; MacKerell, A.D.; Shapiro, P.; Fletcher, S. Small-molecule inhibitors of the ERK signaling pathway: Towards novel anticancer therapeutics. ChemMedChem 2011, 6, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS variant signalling. Biochem. Soc. Trans. 2018, 46, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Fransén, K.; Klintenäs, M.; Osterström, A.; Dimberg, J.; Monstein, H.-J.; Söderkvist, P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis 2004, 25, 527–533. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Tong, J.; Jha, A.; Risnik, D.; Lizardo, D.; Lu, X.; Goel, A.; Opresko, P.L.; Yu, J.; Zhang, L. Synthetical lethality of Werner helicase and mismatch repair deficiency is mediated by p53 and PUMA in colon cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2211775119. [Google Scholar] [CrossRef]
- Morsi, R.Z.; Hage-Sleiman, R.; Kobeissy, H.; Dbaibo, G. Noxa: Role in Cancer Pathogenesis and Treatment. Curr. Cancer Drug Targets 2018, 18, 914–928. [Google Scholar] [CrossRef]
- Padmakumar, V.C.; Aleem, E.; Berthet, C.; Hilton, M.B.; Kaldis, P. Cdk2 and Cdk4 activities are dispensable for tumorigenesis caused by the loss of p53. Mol. Cell. Biol. 2009, 29, 2582–2593. [Google Scholar] [CrossRef] [PubMed]
- Rother, K.; Kirschner, R.; Sänger, K.; Böhlig, L.; Mössner, J.; Engeland, K. p53 downregulates expression of the G1/S cell cycle phosphatase Cdc25A. Oncogene 2007, 26, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Mao, Y.; Zhao, L.; Li, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It′s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.V.; Khromova, N.V.; Kopnin, P.B. Components of the Hepatocellular Carcinoma Microenvironment and Their Role in Tumor Progression. Biochem. Biokhimiia 2017, 82, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, P. Cancer-stromal interactions: Role in cell survival, metabolism and drug sensitivity. Cancer Biol. Ther. 2011, 11, 150–156. [Google Scholar] [CrossRef]
- Kumari, S.; Advani, D.; Sharma, S.; Ambasta, R.K.; Kumar, P. Combinatorial therapy in tumor microenvironment: Where do we stand? Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188585. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [PubMed]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Cupeiro, R.; Pérez-Prieto, R.; Amigo, T.; Gortázar, P.; Redondo, C.; González-Lamuño, D. Role of the monocarboxylate transporter MCT1 in the uptake of lactate during active recovery. Eur. J. Appl. Physiol. 2016, 116, 1005–1010. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Günzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.-Y.; Oh, S.; Kang, Y.K.; Lee, K.-M.; Kang, M.; Jang, Y.J.; Yang, S.-J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Zhai, L.; Zhang, T.; Yin, H.; Gao, H.; Zhao, F.; Wang, Z.; Yang, X.; Jin, M.; et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell 2023, 187, 294–311.e21. [Google Scholar] [CrossRef]
- Harmon, C.; Robinson, M.W.; Hand, F.; Almuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2019, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pyaram, K.; Yarosz, E.L.; Hong, H.; Lyssiotis, C.A.; Giri, S.; Chang, C.-H. Enhanced oxidative phosphorylation in NKT cells is essential for their survival and function. Proc. Natl. Acad. Sci. USA 2019, 116, 7439–7448. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 376–386. [Google Scholar]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Venmar, K.T.; Kimmel, D.W.; Cliffel, D.E.; Fingleton, B. IL4 receptor α mediates enhanced glucose and glutamine metabolism to support breast cancer growth. Biochim. Biophys. Acta 2015, 1853, 1219–1228. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Jethwa, N.; Chung, G.H.C.; Lete, M.G.; Alonso, A.; Byrne, R.D.; Calleja, V.; Larijani, B. Endomembrane PtdIns(3,4,5)P3 activates the PI3K-Akt pathway. J. Cell Sci. 2015, 128, 3456–3465. [Google Scholar] [CrossRef]
- Wang, C.; Yosef, N.; Gaublomme, J.; Wu, C.; Lee, Y.; Clish, C.B.; Kaminski, J.; Xiao, S.; Meyer Zu Horste, G.; Pawlak, M.; et al. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 2015, 163, 1413–1427. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef] [PubMed]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Saieva, L.; Corrado, C.; Fontana, S.; Flugy, A.; Rizzo, A.; De Leo, G.; Alessandro, R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun. Signal. CCS 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Demory Beckler, M.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.-J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell. Proteom. MCP 2013, 12, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef]
- Nishio, M.; Endo, T.; Tsukada, N.; Ohata, J.; Kitada, S.; Reed, J.C.; Zvaifler, N.J.; Kipps, T.J. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood 2005, 106, 1012–1020. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Dicarlo, M.; Bianchi, N.; Ferretti, C.; Orciani, M.; Di Primio, R.; Mattioli-Belmonte, M. Evidence Supporting a Paracrine Effect of IGF-1/VEGF on Human Mesenchymal Stromal Cell Commitment. Cells Tissues Organs 2016, 201, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Hager, J.H.; Ferrara, N.; Gerber, H.-P.; Hanahan, D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 2002, 1, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Bruchim, I. IGF-1 and BRCA1 signalling pathways in familial cancer. Lancet Oncol. 2012, 13, e537–e544. [Google Scholar] [CrossRef] [PubMed]
- Iams, W.T.; Lovly, C.M. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Flashner-Abramson, E.; Shalapour, S.; Zhong, Z.; Taniguchi, K.; Levitzki, A.; Karin, M. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene 2016, 35, 2634–2644. [Google Scholar] [CrossRef] [PubMed]
- Ogata, A.; Chauhan, D.; Teoh, G.; Treon, S.P.; Urashima, M.; Schlossman, R.L.; Anderson, K.C. IL-6 triggers cell growth via the Ras-dependent mitogen-activated protein kinase cascade. J. Immunol. 1997, 159, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.-W.; Kopantzev, E.; Rudikoff, S. Insulinlike growth factor-I signaling in multiple myeloma: Downstream elements, functional correlates, and pathway cross-talk. Blood 2002, 99, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Jewell, A.P.; Worman, C.P.; Lydyard, P.M.; Yong, K.L.; Giles, F.J.; Goldstone, A.H. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br. J. Haematol. 1994, 88, 268–274. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Menzel, T.; Lynen, S.; Wrazel, L.; Rosén, A.; Al-Katib, A.; Raveche, E.; Gabrilove, J.L. Basic fibroblast growth factor (bFGF) upregulates the expression of bcl-2 in B cell chronic lymphocytic leukemia cell lines resulting in delaying apoptosis. Leukemia 1997, 11, 258–265. [Google Scholar] [CrossRef]
- Levesque, M.C.; Misukonis, M.A.; O’Loughlin, C.W.; Chen, Y.; Beasley, B.E.; Wilson, D.L.; Adams, D.J.; Silber, R.; Weinberg, J.B. IL-4 and interferon gamma regulate expression of inducible nitric oxide synthase in chronic lymphocytic leukemia cells. Leukemia 2003, 17, 442–450. [Google Scholar] [CrossRef]
- Sato, A.; Rahman, N.I.A.; Shimizu, A.; Ogita, H. Cell-to-cell contact-mediated regulation of tumor behavior in the tumor microenvironment. Cancer Sci. 2021, 112, 4005–4012. [Google Scholar] [CrossRef]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and nectin-like molecules: Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Liu, S.; Chen, S.; Yuan, W.; Wang, H.; Chen, K.; Li, D.; Li, D. PD-1/PD-L1 interaction up-regulates MDR1/P-gp expression in breast cancer cells via PI3K/AKT and MAPK/ERK pathways. Oncotarget 2017, 8, 99901–99912. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Ann. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, S.F.; Fleming, J.V.; Azevedo, J.E.; Carmo-Fonseca, M.; de Sousa, M. Stimulation of an unfolded protein response impairs MHC class I expression. J. Immunol. 2007, 178, 3612–3619. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Harnoss, J.M.; Le Thomas, A.; Reichelt, M.; Guttman, O.; Wu, T.D.; Marsters, S.A.; Shemorry, A.; Lawrence, D.A.; Kan, D.; Segal, E.; et al. IRE1α Disruption in Triple-Negative Breast Cancer Cooperates with Antiangiogenic Therapy by Reversing ER Stress Adaptation and Remodeling the Tumor Microenvironment. Cancer Res. 2020, 80, 2368–2379. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, Y.-X.; Liu, F.; Sang, L.; Shi, C.; Xie, S.; Bian, W.; Yang, J.-C.; Yang, Z.; Qu, L.; et al. lncRNA BREA2 promotes metastasis by disrupting the WWP2-mediated ubiquitination of Notch1. Proc. Natl. Acad. Sci. USA 2023, 120, e2206694120. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Wang, R.; Chen, Q.; Luo, J.; Wang, J.; Zhao, Z.; Li, Y.; Wang, Y.; Wang, X.; Cheng, B. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 2018, 8, 302–316. [Google Scholar] [PubMed]
- Colombo, M.; Galletti, S.; Bulfamante, G.; Falleni, M.; Tosi, D.; Todoerti, K.; Lazzari, E.; Crews, L.A.; Jamieson, C.H.M.; Ravaioli, S.; et al. Multiple myeloma-derived Jagged ligands increases autocrine and paracrine interleukin-6 expression in bone marrow niche. Oncotarget 2016, 7, 56013–56029. [Google Scholar] [CrossRef]
- Su, Q.; Zhang, B.; Zhang, L.; Dang, T.; Rowley, D.; Ittmann, M.; Xin, L. Jagged1 upregulation in prostate epithelial cells promotes formation of reactive stroma in the Pten null mouse model for prostate cancer. Oncogene 2017, 36, 618–627. [Google Scholar] [CrossRef]
- Janghorban, M.; Yang, Y.; Zhao, N.; Hamor, C.; Nguyen, T.M.; Zhang, X.H.F.; Rosen, J.M. Single-Cell Analysis Unveils the Role of the Tumor Immune Microenvironment and Notch Signaling in Dormant Minimal Residual Disease. Cancer Res. 2022, 82, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, D.-A.; Guan, L.; Myint, P.K.; Chin, L.; Dang, H.; Xu, Y.; Ren, J.; Li, T.; Yu, Z.; et al. Stiff matrix induces exosome secretion to promote tumour growth. Nat. Cell Biol. 2023, 25, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, W.; Wu, R.; Li, J.; Park, S.-A.; Tu, E.; Zanvit, P.; Xu, J.; Liu, O.; Cain, A.; et al. High Glucose Intake Exacerbates Autoimmunity through Reactive-Oxygen-Species-Mediated TGF-β Cytokine Activation. Immunity 2019, 51, 671–681.e5. [Google Scholar] [CrossRef]
- Lawless, S.J.; Kedia-Mehta, N.; Walls, J.F.; McGarrigle, R.; Convery, O.; Sinclair, L.V.; Navarro, M.N.; Murray, J.; Finlay, D.K. Glucose represses dendritic cell-induced T cell responses. Nat. Commun. 2017, 8, 15620. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yan, J.; Liu, Y.; Shi, J.; Wang, H.; Zhou, H.; Zhou, Y.; Zhang, T.; Zhao, L.; Meng, X.; et al. Cancer-cell-derived fumarate suppresses the anti-tumor capacity of CD8+ T cells in the tumor microenvironment. Cell Metab. 2023, 35, 961–978.e10. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.H.; Elia, I.; Shahid, O.; Gaudiano, E.F.; Sifnugel, N.E.; Johnson, S.; Reynolds, A.G.; Fung, M.E.; Joshi, S.; LaFleur, M.W.; et al. Formate Supplementation Enhances Antitumor CD8+ T-cell Fitness and Efficacy of PD-1 Blockade. Cancer Discov. 2023, 13, 2566–2583. [Google Scholar] [CrossRef]
- Tsai, Y.-L.; Arias-Badia, M.; Kadlecek, T.A.; Lwin, Y.M.; Srinath, A.; Shah, N.H.; Wang, Z.-E.; Barber, D.; Kuriyan, J.; Fong, L.; et al. TCR signaling promotes formation of an STS1-Cbl-b complex with pH-sensitive phosphatase activity that suppresses T cell function in acidic environments. Immunity 2023, 56, 2682–2698.e9. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Yan, S.; Wan, G. Tumor-associated macrophages in immunotherapy. FEBS J. 2021, 288, 6174–6186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wei, S.; Dai, S.; Li, X.; Wang, H.; Zhang, H.; Sun, G.; Shan, B.; Zhao, L. The NR_109/FUBP1/c-Myc axis regulates TAM polarization and remodels the tumor microenvironment to promote cancer development. J. Immunother. Cancer 2023, 11, e006230. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Bao, X.; Zou, Y.; Wang, L.; Li, Y.; Yang, L.; Liao, A.; Zhang, X.; Jiang, X.; Liang, D.; et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci. Adv. 2023, 9, eadg2697. [Google Scholar] [CrossRef]
- Yuan, D.; Hu, J.; Ju, X.; Putz, E.M.; Zheng, S.; Koda, S.; Sun, G.; Deng, X.; Xu, Z.; Nie, W.; et al. NMDAR antagonists suppress tumor progression by regulating tumor-associated macrophages. Proc. Natl. Acad. Sci. USA 2023, 120, e2302126120. [Google Scholar] [CrossRef]
- Mao, Y.; Shi, D.; Li, G.; Jiang, P. Citrulline depletion by ASS1 is required for proinflammatory macrophage activation and immune responses. Mol. Cell 2022, 82, 527–541.e7. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Kim, S.; Hong, B.-J.; Lee, C.-J.; Kim, Y.-E.; Bok, S.; Oh, J.-M.; Gwak, S.-H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef]
- Huang, T.-X.; Tan, X.-Y.; Huang, H.-S.; Li, Y.-T.; Liu, B.-L.; Liu, K.-S.; Chen, X.; Chen, Z.; Guan, X.-Y.; Zou, C.; et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut 2022, 71, 333–344. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ota, T.; Watanabe, M.; Imada, K.; Nomizu, M.; Ito, A. Identification of an active site of EMMPRIN for the augmentation of matrix metalloproteinase-1 and -3 expression in a co-culture of human uterine cervical carcinoma cells and fibroblasts. Gynecol. Oncol. 2009, 114, 337–342. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 2016, 48, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441–65453. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Wei, X.-H.; Zhan, X.-M.; He, J.-Y.; Zeng, Y.-Q.; Tian, X.-M.; Yuan, S.-T.; Sun, L. Adipocyte-Derived Leptin Promotes PAI-1 -Mediated Breast Cancer Metastasis in a STAT3/miR-34a Dependent Manner. Cancers 2020, 12, 3864. [Google Scholar] [CrossRef]
- Huang, C.-K.; Chang, P.-H.; Kuo, W.-H.; Chen, C.-L.; Jeng, Y.-M.; Chang, K.-J.; Shew, J.-Y.; Hu, C.-M.; Lee, W.-H. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via β-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Ma, H.; Gao, Y.; Liang, Y.; Du, Y.; Hao, S.; Ni, T. The Tumor Microenvironment: Signal Transduction. Biomolecules 2024, 14, 438. https://doi.org/10.3390/biom14040438
Zhang X, Ma H, Gao Y, Liang Y, Du Y, Hao S, Ni T. The Tumor Microenvironment: Signal Transduction. Biomolecules. 2024; 14(4):438. https://doi.org/10.3390/biom14040438
Chicago/Turabian StyleZhang, Xianhong, Haijun Ma, Yue Gao, Yabing Liang, Yitian Du, Shuailin Hao, and Ting Ni. 2024. "The Tumor Microenvironment: Signal Transduction" Biomolecules 14, no. 4: 438. https://doi.org/10.3390/biom14040438
APA StyleZhang, X., Ma, H., Gao, Y., Liang, Y., Du, Y., Hao, S., & Ni, T. (2024). The Tumor Microenvironment: Signal Transduction. Biomolecules, 14(4), 438. https://doi.org/10.3390/biom14040438