

The Impact of Inorganic Systems and Photoactive Metal Compounds on Cytochrome P450 Enzymes and Metabolism: From Induction to Inhibition

Abstract
1. Introduction
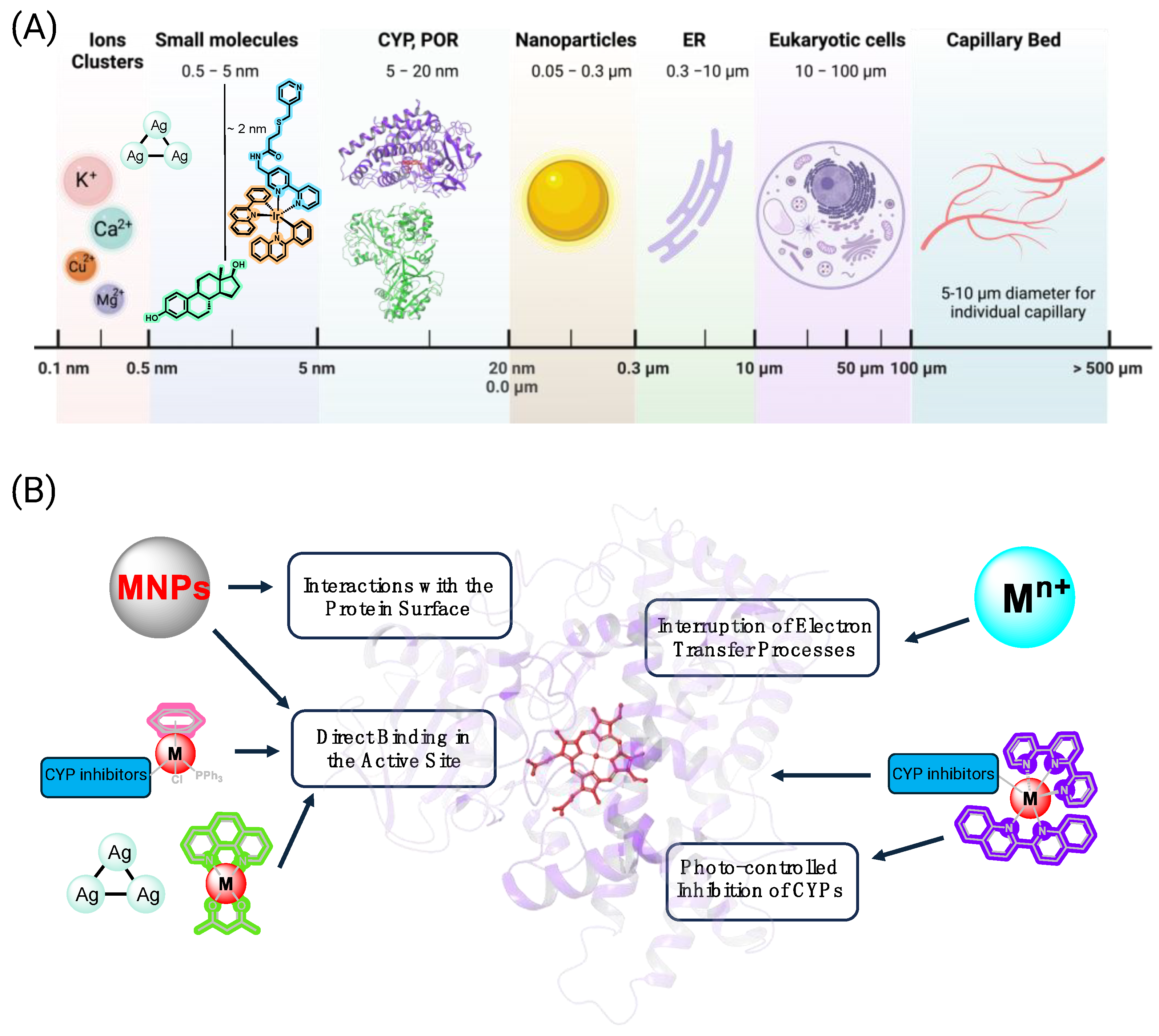
2. Metallic Nanoparticles, Metal Clusters, and Metal Ions
3. Coordination Complexes and Organometallic Compounds
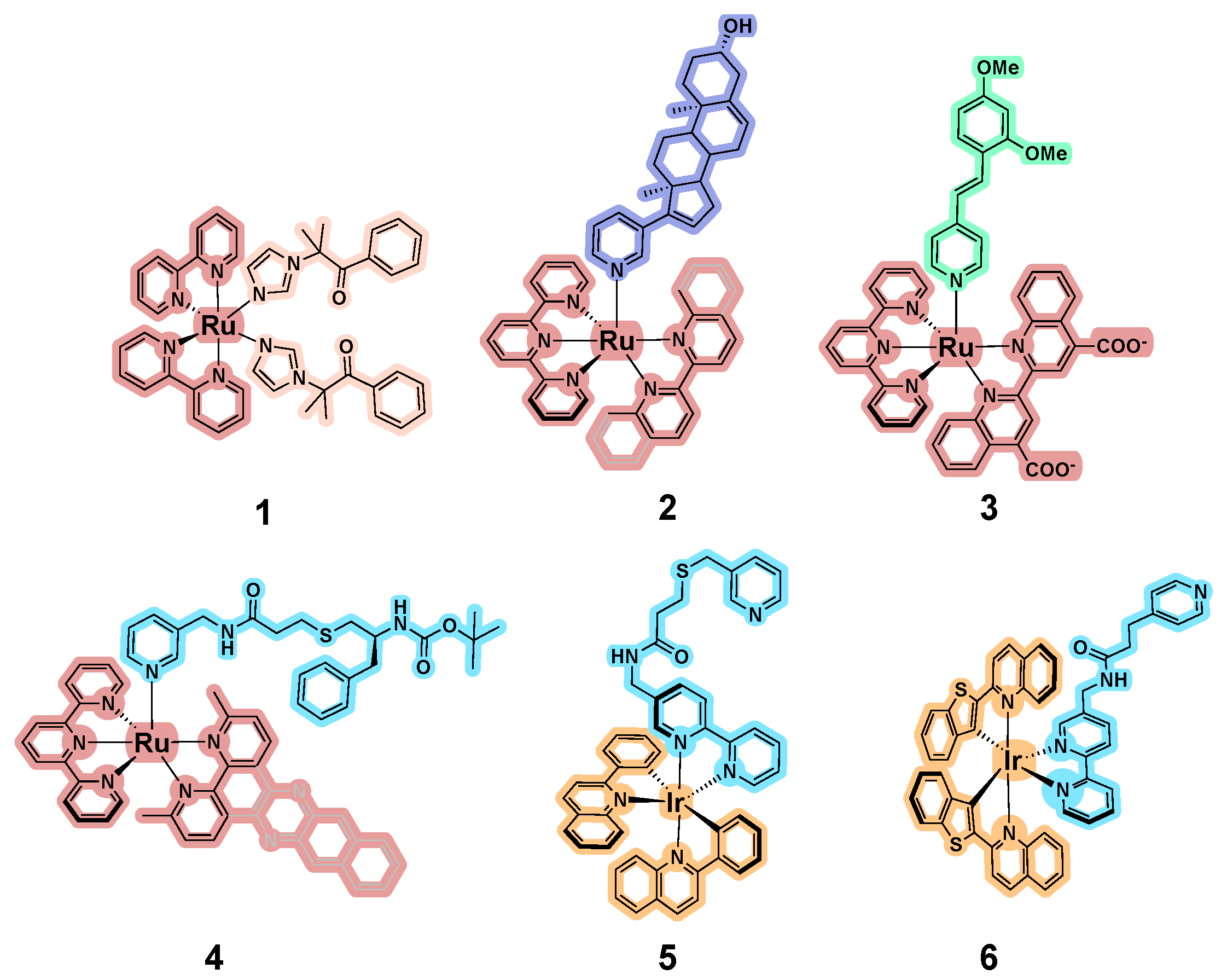
3.1. Development of Inorganic Precursors for Photo-Controlled Inhibition of CYPs
3.2. Binding of Metal-Containing “Wires” with P450 Proteins
3.3. Photostable Inorganic Compounds Bearing Coordinated CYP Inhibitors
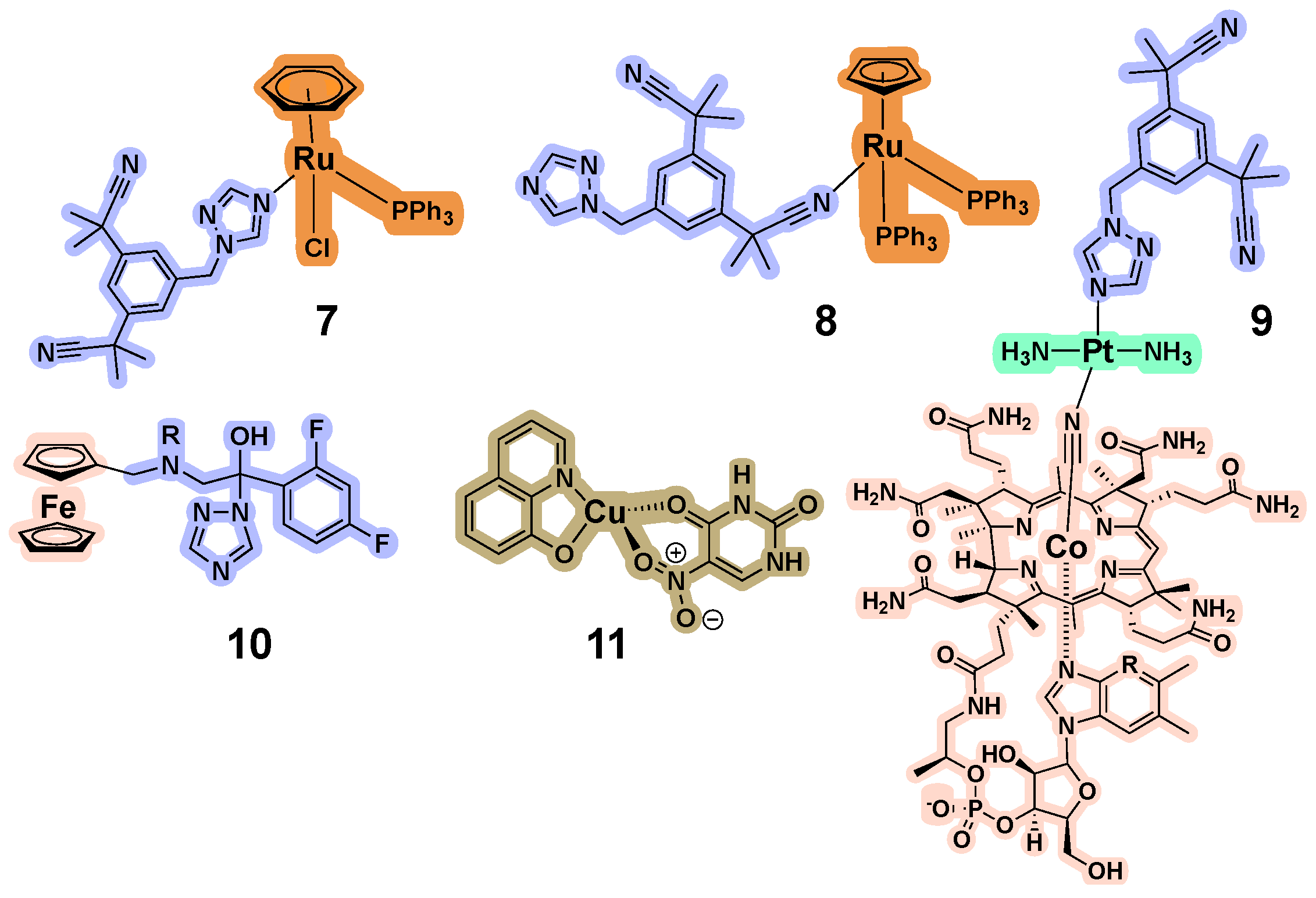
3.4. CYP Inhibition by Metal-Containing Compounds
3.5. Impact of Stability of Metal-Containing Compounds on CYP Inhibition
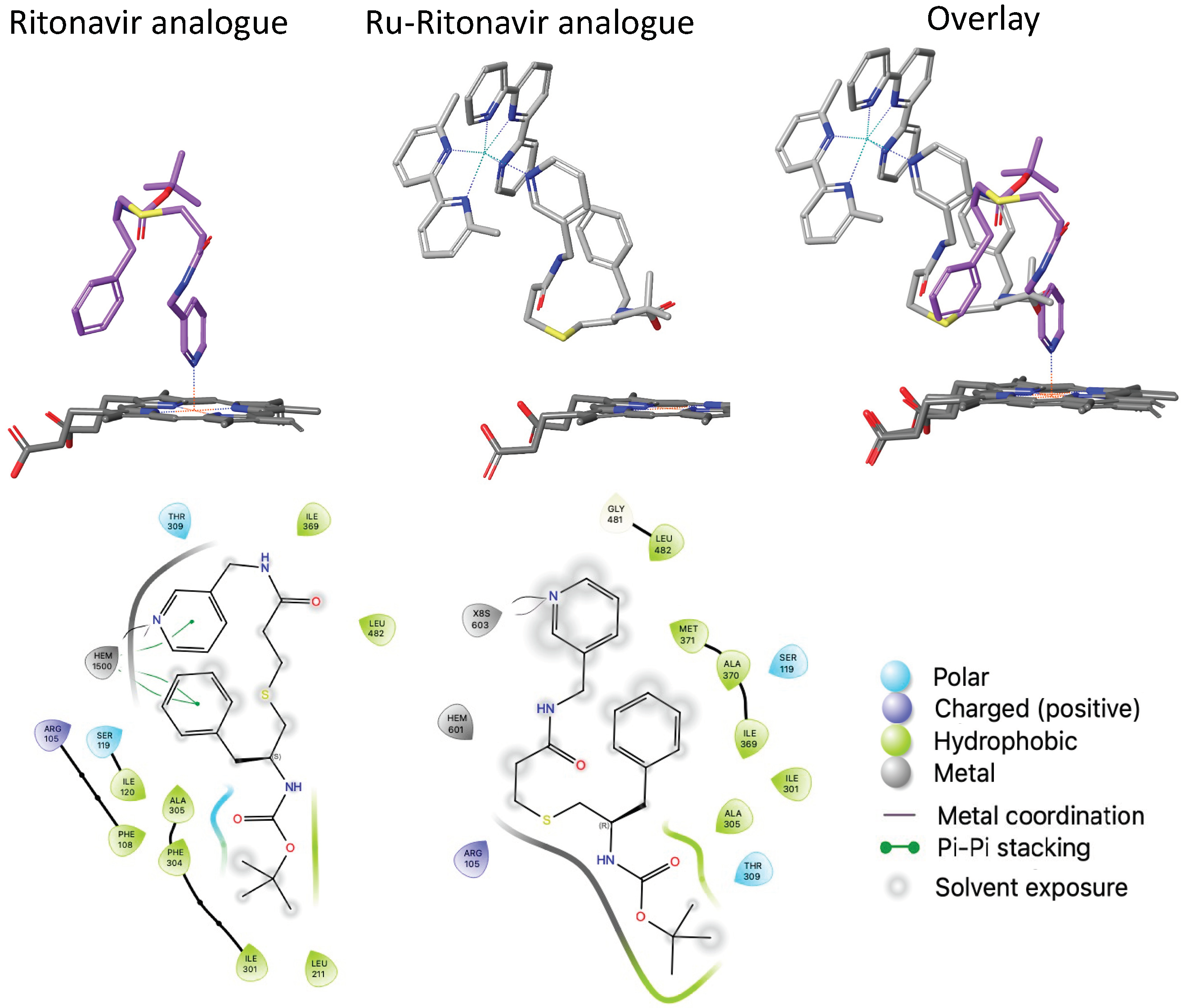
3.6. Structural Studies to Elucidate the Interactions between Inorganic Compounds and CYPs
3.7. Metabolism of Inorganic Drugs by Liver CYPs
3.8. Induction of CYPs by Metals, Metal-Containing Compounds and Drugs, and Enhanced Toxicity
4. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Guengerich, F.P. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J. 2006, 8, E101–E111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Human drug metabolising cytochrome P450 enzymes: Properties and polymorphisms. Naunyn Schmiedeberg’s Arch. Pharmacol. 2004, 369, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Zordoky, B.N.M.; El-Kadi, A.O.S. Effect of cytochrome P450 polymorphism on arachidonic acid metabolism and their impact on cardiovascular diseases. Pharmacol. Ther. 2010, 125, 446–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. A history of the roles of cytochrome P450 enzymes in the toxicity of drugs. Toxicol. Res. 2021, 37, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, E.; Liu, D.; Kallio, A.; Mäkilä, E.; Zhang, H.; Salonen, J.; Santos, H.A.; Sikanen, T.M. The impact of porous silicon nanoparticles on human cytochrome P450 metabolism in human liver microsomes in vitro. Eur. J. Pharm. Sci. 2017, 104, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Kueznik, T.; Samberger, C.; Roblegg, E.; Wrighton, C.; Pieber, T.R. Size-dependent effects of nanoparticles on the activity of cytochrome P450 isoenzymes. Toxicol. Appl. Pharmacol. 2010, 242, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zang, Y.; Qu, J.; Tang, M.; Zhang, T. The Toxicity of Metallic Nanoparticles on Liver: The Subcellular Damages, Mechanisms, and Outcomes. Int. J. Nanomed. 2019, 14, 8787–8804. [Google Scholar] [CrossRef]
- Kulthong, K.; Maniratanachote, R.; Kobayashi, Y.; Fukami, T.; Yokoi, T. Effects of silver nanoparticles on rat hepatic cytochrome P450 enzyme activity. Xenobiotica 2012, 42, 854–862. [Google Scholar] [CrossRef]
- Lamb, J.G.; Hathaway, L.B.; Munger, M.A.; Raucy, J.L.; Franklin, M.R. Nanosilver Particle Effects on Drug Metabolism In Vitro. Drug Metab. Dispos. 2010, 38, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Wasukan, N.; Kuno, M.; Maniratanachote, R. Molecular Docking as a Promising Predictive Model for Silver Nanoparticle-Mediated Inhibition of Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2019, 59, 5126–5134. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Joo, H. Assessment of Gold Nanoparticles-Inhibited Cytochrome P450 3A4 Activity and Molecular Mechanisms Underlying Its Cellular Toxicity in Human Hepatocellular Carcinoma Cell Line C3A. Nanoscale Res. Lett. 2018, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Tang, L.; Luo, M.; Zhou, J.; Guo, B.; Liu, Y.; Chen, B. Size- and time-dependent alteration in metabolic activities of human hepatic cytochrome P450 isozymes by gold nanoparticles via microsomal coincubations. Nanoscale Res. Lett. 2014, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xu, M.; Shi, F.; Ye, G.; Lv, C.; Luo, J.; Zhao, L.; Li, Y. Effects and Mechanism of Nano-Copper Exposure on Hepatic Cytochrome P450 Enzymes in Rats. Int. J. Mol. Sci. 2018, 19, 2140. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Xu, M.; Luo, J.; Zhao, L.; Shi, F.; Ye, G.; Lv, C.; Li, Y. Effect of copper nanoparticles on brain cytochrome P450 enzymes in rats. Mol. Med. Rep. 2019, 20, 771–778. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, M.; Deng, Y.; Li, H.; Bu, Q.; Liu, R.; Yu, J.; Liu, S.; Zeng, Z.; Sun, W.; et al. Copper nanoparticles lead to reproductive dysfunction by affecting key enzymes of ovarian hormone synthesis and metabolism in female rats. Ecotoxicol. Environ. Saf. 2023, 254, 114704. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Zhang, X.; Yu, Q.; Liu, L.; Zhou, X. Cytochrome P450-dependent reactive oxygen species (ROS) production contributes to Mn3O4 nanoparticle-caused liver injury. RSC Adv. 2018, 8, 37307–37314. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ong, C.E.; Pung, Y.F.; Chieng, J.Y. The current understanding of the interactions between nanoparticles and cytochrome P450 enzymes—A literature-based review. Xenobiotica 2019, 49, 863–876. [Google Scholar] [CrossRef]
- Al-Btoush, H.A. The Interactions between Metallic Nanoparticles and Cytochrome P450, Alanine Aminotransferase, and Aspartate Aminotransferase Enzymes. J. Pure Appl. Microbiol. 2023, 17, 2024–2040. [Google Scholar] [CrossRef]
- Westlake, A.C.G.; Harford-Cross, C.F.; Donovan, J.; Wong, L.L. Mutations of glutamate-84 at the putative potassium-binding site affect camphor binding and oxidation by cytochrome P450. Eur. J. Biochem. 1999, 265, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.K.; Mazumdar, S. Reversible inactivation of cytochrome P450 by alkaline earth metal ions: Auxiliary metal ion induced conformation change and formation of inactive P420 species in CYP101. J. Inorg. Biochem. 2008, 102, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Ahn, T.; Yim, S.-K.; Yun, C.-H. Differential Effect of Copper (II) on the Cytochrome P450 Enzymes and NADPH−Cytochrome P450 Reductase: Inhibition of Cytochrome P450-Catalyzed Reactions by Copper (II) Ion. Biochemistry 2002, 41, 9438–9447. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Yun, C.-H. Inhibition of human cytochrome P450 3A4 activity by zinc(II) ion. Toxicol. Lett. 2005, 156, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.A.; Warwicker, J.; de Visser, S.P. How Do Metal Ions Modulate the Rate-Determining Electron-Transfer Step in Cytochrome P450 Reactions? Chem. Eur. J. 2020, 26, 15270–15281. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Lei, L.; Vassylyev, D.G.; Lin, X.; Cane, D.E.; Kelly, S.L.; Yuan, H.; Lamb, D.C.; Waterman, M.R. Crystal Structure of Albaflavenone Monooxygenase Containing a Moonlighting Terpene Synthase Active Site. J. Biol. Chem. 2009, 284, 36711–36719. [Google Scholar] [CrossRef] [PubMed]
- El-Ghiaty, M.A.; El-Kadi, A.O.S. Arsenic: Various species with different effects on cytochrome P450 regulation in humans. EXCLI J. 2021, 20, 1184–1242. [Google Scholar] [CrossRef] [PubMed]
- Naraharisetti, S.B.; Aggarwal, M.; Sarkar, S.N.; Malik, J.K. Concurrent subacute exposure to arsenic through drinking water and malathion via diet in male rats: Effects on hepatic drug-metabolizing enzymes. Arch. Toxicol. 2008, 82, 543–551. [Google Scholar] [CrossRef]
- Anwar-Mohamed, A.; El-Sherbeni, A.; Kim, S.H.; Elshenawy, O.H.; Althurwi, H.N.; Zordoky, B.N.; El-Kadi, A.O. Acute arsenic treatment alters cytochrome P450 expression and arachidonic acid metabolism in lung, liver and kidney of C57Bl/6 mice. Xenobiotica 2013, 43, 719–729. [Google Scholar] [CrossRef]
- Terrones-Gurrola, M.; Ponce-Peña, P.; Salas-Pacheco, J.M.; Camacho-Luis, A.; Pozos-Guillén, A.J.; Nieto-Delgado, G.; López-Guzmán, O.D.; Vértiz-Hernández, A.A. Arsenic: A Perspective on Its Effect on Pioglitazone Bioavailability. Int. J. Environ. Res. Public Health 2023, 20, 1901. [Google Scholar] [CrossRef]
- Ener, M.E.; Lee, Y.T.; Winkler, J.R.; Gray, H.B.; Cheruzel, L. Photooxidation of cytochrome P450-BM3. Proc. Natl. Acad. Sci. USA 2010, 107, 18783–18786. [Google Scholar] [CrossRef] [PubMed]
- Dwaraknath, S.; Tran, N.-H.; Dao, T.; Colbert, A.; Mullen, S.; Nguyen, A.; Cortez, A.; Cheruzel, L. A facile and versatile methodology for cysteine specific labeling of proteins with octahedral polypyridyl d6 metal complexes. J. Inorg. Biochem. 2014, 136, 154–160. [Google Scholar] [CrossRef]
- Tran, N.-H.; Nguyen, D.; Dwaraknath, S.; Mahadevan, S.; Chavez, G.; Nguyen, A.; Dao, T.; Mullen, S.; Nguyen, T.-A.; Cheruzel, L.E. An Efficient Light-Driven P450 BM3 Biocatalyst. J. Am. Chem. Soc. 2013, 135, 14484–14487. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.-H.; Huynh, N.; Bui, T.; Nguyen, Y.; Huynh, P.; Cooper, M.E.; Cheruzel, L.E. Light-initiated hydroxylation of lauric acid using hybrid P450 BM3 enzymes. Chem. Commun. 2011, 47, 11936–11938. [Google Scholar] [CrossRef]
- Kato, M.; Nguyen, D.; Gonzalez, M.; Cortez, A.; Mullen, S.E.; Cheruzel, L.E. Regio- and stereoselective hydroxylation of 10-undecenoic acid with a light-driven P450 BM3 biocatalyst yielding a valuable synthon for natural product synthesis. Bioorganic Med. Chem. 2014, 22, 5687–5691. [Google Scholar] [CrossRef] [PubMed]
- Do, M.Q.; Henry, E.; Kato, M.; Cheruzel, L. Cross-linked cytochrome P450 BM3 aggregates promoted by Ru(II)-diimine complexes bearing aldehyde groups. J. Inorg. Biochem. 2018, 186, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Foley, B.; Vu, J.; Huynh, M.; Lucero, K.; Harmon, C.; Cheruzel, L. Promoting P450 BM3 heme domain dimerization with a tris(5-iodoacetamido-1,10-phenanthroline)Ru(II) complex. Biotechnol. Appl. Biochem. 2020, 67, 536–540. [Google Scholar] [CrossRef]
- Eidenschenk, C.; Cheruzel, L. Ru(II)-diimine complexes and cytochrome P450 working hand-in-hand. J. Inorg. Biochem. 2020, 213, 111254. [Google Scholar] [CrossRef]
- Lincoln, S.F. Mechanistic Studies of Metal Aqua Ions: A Semi-Historical Perspective. Helv. Chim. Acta 2005, 88, 523–545. [Google Scholar] [CrossRef]
- Zamora, A.; Denning, C.A.; Heidary, D.K.; Wachter, E.; Nease, L.A.; Ruiz, J.; Glazer, E.C. Ruthenium-containing P450 inhibitors for dual enzyme inhibition and DNA damage. Dalton Trans. 2017, 46, 2165–2173. [Google Scholar] [CrossRef]
- Miura, Y.; Fulco, A.J. Omega-1, Omega-2 and Omega-3 hydroxylation of long-chain fatty acids, amides and alcohols by a soluble enzyme system from Bacillus megaterium. Biochim. Biophys. Acta 1975, 388, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, G.; Gilardi, G. Optimization of the bacterial cytochrome P450 BM3 system for the production of human drug metabolites. Int. J. Mol. Sci. 2012, 13, 15901–15924. [Google Scholar] [CrossRef]
- Damsten, M.C.; van Vugt-Lussenburg, B.M.A.; Zeldenthuis, T.; de Vlieger, J.S.B.; Commandeur, J.N.M.; Vermeulen, N.P.E. Application of drug metabolising mutants of cytochrome P450 BM3 (CYP102A1) as biocatalysts for the generation of reactive metabolites. Chem.-Biol. Interact. 2008, 171, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Reinen, J.; van Leeuwen, J.S.; Li, Y.; Sun, L.; Grootenhuis, P.D.; Decker, C.J.; Saunders, J.; Vermeulen, N.P.; Commandeur, J.N. Efficient screening of cytochrome P450 BM3 mutants for their metabolic activity and diversity toward a wide set of drug-like molecules in chemical space. Drug Metab. Dispos. 2011, 39, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, Y.; Osawa, Y.; Chen, Y.E.; Zhang, H. Computational redesign of cytochrome P450 CYP102A1 for highly stereoselective omeprazole hydroxylation by UniDesign. J. Biol. Chem. 2023, 299, 105050. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; Leys, D.G.; McLean, K.J.; Marshall, K.R.; Ost, T.W.B.; Daff, S.; Miles, C.S.; Chapman, S.K.; Lysek, D.A.; Moser, C.C.; et al. P450 BM3: The very model of a modern flavocytochrome. Trends Biochem. Sci. 2002, 27, 250–257. [Google Scholar] [CrossRef]
- Li, A.; Yadav, R.; White, J.K.; Herroon, M.K.; Callahan, B.P.; Podgorski, I.; Turro, C.; Scott, E.E.; Kodanko, J.J. Illuminating cytochrome P450 binding: Ru(ii)-caged inhibitors of CYP17A1. Chem. Commun. 2017, 53, 3673–3676. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Hachey, A.C.; Fenton, A.; Heidary, D.K.; Glazer, E.C. Ru(II) photocages enable precise control over enzyme activity with red light. Nat. Commun. 2022, 13, 3636. [Google Scholar] [CrossRef] [PubMed]
- Toupin, N.; Steinke, S.J.; Nadella, S.; Li, A.; Rohrabaugh, T.N., Jr.; Samuels, E.R.; Turro, C.; Sevrioukova, I.F.; Kodanko, J.J. Photosensitive Ru(II) Complexes as Inhibitors of the Major Human Drug Metabolizing Enzyme CYP3A4. J. Am. Chem. Soc. 2021, 143, 9191–9205. [Google Scholar] [CrossRef]
- Wachter, E.; Heidary, D.K.; Howerton, B.S.; Parkin, S.; Glazer, E.C. Light-activated ruthenium complexes photobind DNA and are cytotoxic in the photodynamic therapy window. Chem. Commun. 2012, 48, 9649–9651. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Stevens, K.; Parkin, S.; Glazer, E.C. Toward Optimal Ru(II) Photocages: Balancing Photochemistry, Stability, and Biocompatibility Through Fine Tuning of Steric, Electronic, and Physiochemical Features. Inorg. Chem. 2020, 59, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Dmochowski, I.J.; Crane, B.R.; Wilker, J.J.; Winkler, J.R.; Gray, H.B. Optical detection of cytochrome P450 by sensitizer-linked substrates. Proc. Nat. Acad. Sci. USA 1999, 96, 12987–12990. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.R.; Dmochowski, I.J.; Bilwes, A.M.; Gray, H.B.; Crane, B.R. Probing the open state of cytochrome P450cam with ruthenium-linker substrates. Proc. Natl. Acad. Sci. USA 2001, 98, 12420–12425. [Google Scholar] [CrossRef] [PubMed]
- Gorren, A.C.; Mayer, B. Nitric-oxide synthase: A cytochrome P450 family foster child. Biochim. Biophys. Acta 2007, 1770, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.T.; Skubic, C.; Lee, C.M.; Cokan, K.B.; Rozman, D. Regulation of cytochrome P450 enzyme activity and expression by nitric oxide in the context of inflammatory disease. Drug Metab. Rev. 2020, 52, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Belliston-Bittner, W.; Dunn, A.R.; Nguyen, Y.H.L.; Stuehr, D.J.; Winkler, J.R.; Gray, H.B. Picosecond Photoreduction of Inducible Nitric Oxide Synthase by Rhenium(I)−Diimine Wires. J. Am. Chem. Soc. 2005, 127, 15907–15915. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nguyen, Y.H.L.; Winkler, J.R.; Gray, H.B. Probing Heme Coordination States of Inducible Nitric Oxide Synthase with a Re(I)(imidazole-alkyl-nitroarginine) Sensitizer-Wire. J. Phys. Chem. B 2007, 111, 6628–6633. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Glazer, E.C.; Nguyen, Y.H.; Gray, H.B.; Goodin, D.B. Probing inducible nitric oxide synthase with a pterin-ruthenium(II) sensitizer wire. Angew. Chem. Int. Ed. Engl. 2008, 47, 898–901. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Whited, C.A.; Belliston-Bittner, W.; Dunn, A.R.; Winkler, J.R.; Gray, H.B. Probing the heme-thiolate oxygenase domain of inducible nitric oxide synthase with Ru(II) and Re(I) electron tunneling wires. J. Porphyr. Phthalocya. 2008, 12, 971–978. [Google Scholar] [CrossRef]
- Whited, C.A.; Belliston-Bittner, W.; Dunn, A.R.; Winkler, J.R.; Gray, H.B. Nanosecond photoreduction of inducible nitric oxide synthase by a Ru-diimine electron tunneling wire bound distant from the active site. J. Inorg. Biochem. 2009, 103, 906–911. [Google Scholar] [CrossRef][Green Version]
- Denison, M.; Steinke, S.J.; Majeed, A.; Turro, C.; Kocarek, T.A.; Sevrioukova, I.F.; Kodanko, J.J. Ir(III)-Based Agents for Monitoring the Cytochrome P450 3A4 Active Site Occupancy. Inorg. Chem. 2022, 61, 13673–13677. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.; Ahrens, J.J.; Dunbar, M.N.; Warmahaye, H.; Majeed, A.; Turro, C.; Kocarek, T.A.; Sevrioukova, I.F.; Kodanko, J.J. Dynamic Ir(III) Photosensors for the Major Human Drug-Metabolizing Enzyme Cytochrome P450 3A4. Inorg. Chem. 2023, 62, 3305–3320. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinković, D.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Wester, M.R.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J. Biol. Chem. 2004, 279, 38091–38094. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.R.; Hays, A.-M.A.; Goodin, D.B.; Stout, C.D.; Chiu, R.; Winkler, J.R.; Gray, H.B. Fluorescent Probes for Cytochrome P450 Structural Characterization and Inhibitor Screening. J. Am. Chem. Soc. 2002, 124, 10254–10255. [Google Scholar] [CrossRef] [PubMed]
- Hays, A.M.; Dunn, A.R.; Chiu, R.; Gray, H.B.; Stout, C.D.; Goodin, D.B. Conformational states of cytochrome P450cam revealed by trapping of synthetic molecular wires. J. Mol. Biol. 2004, 344, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-T.; Glazer, E.C.; Wilson, R.F.; Stout, C.D.; Goodin, D.B. Three Clusters of Conformational States in P450cam Reveal a Multistep Pathway for Closing of the Substrate Access Channel. Biochemistry 2011, 50, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, R.; Shao, Q.; Wu, Z.; Cui, J.; Meng, Q.; Li, S. Design and Synthesis of Novel Near-Infrared Fluorescence Probes Based on an Open Conformation of a Cytochrome P450 1B1 Complex for Molecular Imaging of Colorectal Tumors. J. Med. Chem. 2023, 66, 16032–16050. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, D.; Fan, Q.; Wu, Z.; Dong, J.; Cui, J.; Wang, J.; Xu, T.; Meng, Q.; Li, S. Design, Synthesis and In Vivo Fluorescence Imaging Study of a Cytochrome P450 1B1 Targeted NIR Probe Containing a Chelator Moiety. Chembiochem 2022, 23, e202200268. [Google Scholar] [CrossRef]
- Yuan, R.X.; Xiong, R.G.; Abrahams, B.F.; Lee, G.H.; Peng, S.M.; Che, C.M.; You, X.Z. A Cu(I) coordination polymer employing a nonsteroidal aromatase inhibitor letrozole as a building block. J. Chem. Soc. Dalton Trans. 2001, 14, 2071–2073. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Zhou, M.; Huang, J.; Cao, Z.; Qi, T.T.; Huang, G.H.; Wen, H.R. Synthesis, Crystal Structure, and Characterization of three New Letrozole Complexes. Z. Anorg. Allg. Chem. 2012, 638, 372–376. [Google Scholar] [CrossRef]
- Castonguay, A.; Doucet, C.; Juhas, M.; Maysinger, D. New Ruthenium(II)–Letrozole Complexes as Anticancer Therapeutics. J. Med. Chem. 2012, 55, 8799–8806. [Google Scholar] [CrossRef] [PubMed]
- Golbaghi, G.; Haghdoost, M.M.; Yancu, D.; de los Santos, Y.L.; Doucet, N.; Patten, S.A.; Sanderson, J.T.; Castonguay, A. Organoruthenium(II) Complexes Bearing an Aromatase Inhibitor: Synthesis, Characterization, Biological Activity and Toxicity in Zebrafish Embryos. Organometallics 2019, 38, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Golbaghi, G.; Pitard, I.; Lucas, M.; Haghdoost, M.M.; de los Santos, Y.L.; Doucet, N.; Patten, S.A.; Sanderson, J.T.; Castonguay, A. Synthesis and biological assessment of a ruthenium(II) cyclopentadienyl complex in breast cancer cells and on the development of zebrafish embryos. Eur. J. Med. Chem. 2020, 188, 112030. [Google Scholar] [CrossRef] [PubMed]
- Golbaghi, G.; Castonguay, A. Rationally Designed Ruthenium Complexes for Breast Cancer Therapy. Molecules 2020, 25, 265. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.Q.; Furger, E.; Alberto, R. Two-step activation prodrugs: Transplatin mediated binding of chemotherapeutic agents to vitamin B12. Org. Biomol. Chem. 2013, 11, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Cisneros-Fajardo, E.J.; Lehmann, T.; Sánchez-Delgado, R.A.; Atencio, R.; Silva, P.; Lira, R.; Urbina, J.A. Toward a Novel Metal-Based Chemotherapy against Tropical Diseases. 6. Synthesis and Characterization of New Copper(II) and Gold(I) Clotrimazole and Ketoconazole Complexes and Evaluation of Their Activity against Trypanosoma cruzi. Inorg. Chem. 2001, 40, 6879–6884. [Google Scholar] [CrossRef] [PubMed]
- Robles-Escajeda, E.; Martínez, A.; Varela-Ramirez, A.; Sánchez-Delgado, R.A.; Aguilera, R.J. Analysis of the cytotoxic effects of ruthenium-ketoconazole and ruthenium-clotrimazole complexes on cancer cells. Cell Biol. Toxicol. 2013, 29, 431–443. [Google Scholar] [CrossRef]
- Iniguez, E.; Sánchez, A.; Vasquez, M.A.; Martínez, A.; Olivas, J.; Sattler, A.; Sánchez-Delgado, R.A.; Maldonado, R.A. Metal-drug synergy: New ruthenium(II) complexes of ketoconazole are highly active against Leishmania major and Trypanosoma cruzi and nontoxic to human or murine normal cells. J. Biol. Inorg. Chem. 2013, 18, 779–790. [Google Scholar] [CrossRef]
- Gagini, T.; Colina-Vegas, L.; Villarreal, W.; Borba-Santos, L.P.; de Souza Pereira, C.; Batista, A.A.; Kneip Fleury, M.; de Souza, W.; Rozental, S.; Costa, L.A.S.; et al. Metal–azole fungistatic drug complexes as anti-Sporothrix spp. agents. New J. Chem. 2018, 42, 13641–13650. [Google Scholar] [CrossRef]
- Colina-Vegas, L.; Oliveira, K.M.; Cunha, B.N.; Cominetti, M.R.; Navarro, M.; Azevedo Batista, A. Anti-Proliferative and Anti-Migration Activity of Arene–Ruthenium(II) Complexes with Azole Therapeutic Agents. Inorganics 2018, 6, 132. [Google Scholar] [CrossRef]
- Colina-Vegas, L.; Lima Prado Godinho, J.; Coutinho, T.; Correa, R.S.; de Souza, W.; Cola Fernandes Rodrigues, J.; Batista, A.A.; Navarro, M. Antiparasitic activity and ultrastructural alterations provoked by organoruthenium complexes against Leishmania amazonensis. New J. Chem. 2019, 43, 1431–1439. [Google Scholar] [CrossRef]
- de Azevedo-França, J.A.; Borba-Santos, L.P.; de Almeida Pimentel, G.; Franco, C.H.J.; Souza, C.; de Almeida Celestino, J.; de Menezes, E.F.; Dos Santos, N.P.; Vieira, E.G.; Ferreira, A.; et al. Antifungal promising agents of zinc(II) and copper(II) derivatives based on azole drug. J. Inorg. Biochem. 2021, 219, 111401. [Google Scholar] [CrossRef]
- Rubbiani, R.; Weil, T.; Tocci, N.; Mastrobuoni, L.; Jeger, S.; Moretto, M.; Ng, J.; Lin, Y.; Hess, J.; Ferrari, S.; et al. In vivo active organometallic-containing antimycotic agents. RSC Chem. Biol. 2021, 2, 1263–1273. [Google Scholar] [CrossRef]
- Campero-Peredo, C.; Bravo-Gómez, M.E.; Hernández-Ojeda, S.L.; Olguin-Reyes, S.d.R.; Espinosa-Aguirre, J.J.; Ruiz-Azuara, L. Effect of [Cu(4,7-dimethyl-1,10-phenanthroline)(acetylacetonato)]NO3, Casiopeína III-Ea, on the activity of cytochrome P450. Toxicol. Vitr. 2016, 33, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Pingaew, R.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Investigation of aromatase inhibitory activity of metal complexes of β-hydroxyquinoline and uracil derivatives. Drug Des. Dev. Ther. 2014, 8, 1089–1096. [Google Scholar] [CrossRef]
- Medvedikova, M.; Ranc, V.; Vanco, J.; Travnicek, Z.; Anzenbacher, P. Highly Cytotoxic Copper(II) Mixed-Ligand Quinolinonato Complexes: Pharmacokinetic Properties and Interactions with Drug Metabolizing Cytochromes P450. Pharmaceutics 2023, 15, 1314. [Google Scholar] [CrossRef] [PubMed]
- Masek, V.; Anzenbacherová, E.; Machová, M.; Brabec, V.; Anzenbacher, P. Interaction of antitumor platinum complexes with human liver microsomal cytochromes P450. Anticancer Drugs 2009, 20, 305–311. [Google Scholar] [CrossRef]
- Respondek, T.; Garner, R.N.; Herroon, M.K.; Podgorski, I.; Turro, C.; Kodanko, J.J. Light Activation of a Cysteine Protease Inhibitor: Caging of a Peptidomimetic Nitrile with RuII(bpy)2. J. Am. Chem. Soc. 2011, 133, 17164–17167. [Google Scholar] [CrossRef]
- Huisman, M.; White, J.K.; Lewalski, V.G.; Podgorski, I.; Turro, C.; Kodanko, J.J. Caging the uncageable: Using metal complex release for photochemical control over irreversible inhibition. Chem. Commun. 2016, 52, 12590–12593. [Google Scholar] [CrossRef] [PubMed]
- Arora, K.; Herroon, M.; Al-Afyouni, M.H.; Toupin, N.P.; Rohrabaugh, T.N., Jr.; Loftus, L.M.; Podgorski, I.; Turro, C.; Kodanko, J.J. Catch and Release Photosensitizers: Combining Dual-Action Ruthenium Complexes with Protease Inactivation for Targeting Invasive Cancers. J. Am. Chem. Soc. 2018, 140, 14367–14380. [Google Scholar] [CrossRef] [PubMed]
- Lameijer, L.N.; Ernst, D.; Hopkins, S.L.; Meijer, M.S.; Askes, S.H.C.; Le Dévédec, S.E.; Bonnet, S. A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed. 2017, 56, 11549–11553. [Google Scholar] [CrossRef] [PubMed]
- van Rixel, V.H.S.; Ramu, V.; Auyeung, A.B.; Beztsinna, N.; Leger, D.Y.; Lameijer, L.N.; Hilt, S.T.; Le Dévédec, S.E.; Yildiz, T.; Betancourt, T.; et al. Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model. J. Am. Chem. Soc. 2019, 141, 18444–18454. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Carroll, P.J.; Meggers, E. Rapid Access to Unexplored Chemical Space by Ligand Scanning around a Ruthenium Center: Discovery of Potent and Selective Protein Kinase Inhibitors. J. Am. Chem. Soc. 2006, 128, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.L.; Liu, W.F.; Chen, Y.; Luo, H.B.; Sun, L.B.; Chen, X.W.; Zhou, Z.W.; Sneed, K.B.; Li, C.G.; Du, Y.M.; et al. Ligand- and protein-based modeling studies of the inhibitors of human cytochrome P450 2D6 and a virtual screening for potential inhibitors from the Chinese herbal medicine, Scutellaria baicalensis (Huangqin, Baikal Skullcap). Comb. Chem. High. Throughput Screen. 2012, 15, 36–80. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Griffin, K.J.; Song, W.; Richardson, T.H.; Yang, M.; Johnson, E.F. Identification of amino acid substitutions that confer a high affinity for sulfaphenazole binding and a high catalytic efficiency for warfarin metabolism to P450 2C19. Biochemistry 1998, 37, 16270–16279. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Q.; He, Y.A.; Halpert, J.R. Escherichia coli Expression of Site-Directed Mutants of Cytochrome P450 2B1 from Six Substrate Recognition Sites: Substrate Specificity and Inhibitor Selectivity Studies. Chem. Res. Toxicol. 1995, 8, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Tiong, K.H.; Mohammed Yunus, N.A.; Yiap, B.C.; Tan, E.L.; Ismail, R.; Ong, C.E. Inhibitory potency of 8-methoxypsoralen on cytochrome P450 2A6 (CYP2A6) allelic variants CYP2A6 15, CYP2A6 16, CYP2A6 21 and CYP2A6 22: Differential susceptibility due to different sequence locations of the mutations. PLoS ONE 2014, 9, e86230. [Google Scholar] [CrossRef]
- Fowler, S.M.; Taylor, J.M.; Friedberg, T.; Wolf, C.R.; Riley, R.J. CYP3A4 Active Site Volume Modification by Mutagenesis of Leucine 211. Drug Metab. Dispos. 2002, 30, 452–456. [Google Scholar] [CrossRef]
- Fowler, S.M.; Riley, R.J.; Pritchard, M.P.; Sutcliffe, M.J.; Friedberg, T.; Wolf, C.R. Amino acid 305 determines catalytic center accessibility in CYP3A4. Biochemistry 2000, 39, 4406–4414. [Google Scholar] [CrossRef]
- Domanski, T.L.; He, Y.-A.; Khan, K.K.; Roussel, F.; Wang, Q.; Halpert, J.R. Phenylalanine and Tryptophan Scanning Mutagenesis of CYP3A4 Substrate Recognition Site Residues and Effect on Substrate Oxidation and Cooperativity. Biochemistry 2001, 40, 10150–10160. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.K.; He, Y.Q.; Domanski, T.L.; Halpert, J.R. Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: An evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol. Pharmacol. 2002, 61, 495–506. [Google Scholar] [CrossRef]
- Sevrioukova, I.F. High-Level Production and Properties of the Cysteine-Depleted Cytochrome P450 3A4. Biochemistry 2017, 56, 3058–3067. [Google Scholar] [CrossRef]
- Geronimo, I.; Denning, C.A.; Rogers, W.E.; Othman, T.; Huxford, T.; Heidary, D.K.; Glazer, E.C.; Payne, C.M. Effect of Mutation and Substrate Binding on the Stability of Cytochrome P450BM3 Variants. Biochemistry 2016, 55, 3594–3606. [Google Scholar] [CrossRef]
- Geronimo, I.; Denning, C.A.; Heidary, D.K.; Glazer, E.C.; Payne, C.M. Molecular Determinants of Substrate Affinity and Enzyme Activity of a Cytochrome P450BM3 Variant. Biophys. J. 2018, 115, 1251–1263. [Google Scholar] [CrossRef]
- Lampe, J.N.; Brandman, R.; Sivaramakrishnan, S.; de Montellano, P.R.O. Two-dimensional NMR and all-atom molecular dynamics of cytochrome P450 CYP119 reveal hidden conformational substates. J. Biol. Chem. 2010, 285, 9594–9603. [Google Scholar] [CrossRef]
- Hakkennes, M.L.A.; Buda, F.; Bonnet, S. MetalDock: An Open Access Docking Tool for Easy and Reproducible Docking of Metal Complexes. J. Chem. Inf. Model. 2023, 63, 7816–7825. [Google Scholar] [CrossRef] [PubMed]
- Blocka, K. Auranofin versus injectable gold. Comparison of pharmacokinetic properties. Am. J. Med. 1983, 75, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Quintanilha, J.C.F.; de Sousa, V.M.; Visacri, M.B.; Amaral, L.S.; Santos, R.M.M.; Zambrano, T.; Salazar, L.A.; Moriel, P. Involvement of cytochrome P450 in cisplatin treatment: Implications for toxicity. Cancer Chemother. Pharmacol. 2017, 80, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Ong, Y.C.; Lin, Y.; Cariou, K.; Gasser, G. A tutorial for the assessment of the stability of organometallic complexes in biological media. J. Organomet. Chem. 2020, 906, 121059. [Google Scholar] [CrossRef]
- Tompkins, L.M.; Wallace, A.D. Mechanisms of cytochrome P450 induction. J. Biochem. Mol. Toxicol. 2007, 21, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Czekaj, P.; Skowronek, R. Transcription Factors Potentially Involved in Regulation of Cytochrome P450 Gene Expression. In Topics on Drug Metabolism; James, P., Ed.; IntechOpen: Rijeka, Croatia, 2012; Chapter 7. [Google Scholar]
- Wangcharoenrung, L.; Warisnoicharoen, W. Change in mRNA expression of human cytochrome P450 by gold nanoparticles. J. Biol. Sci. 2011, 11, 173–180. [Google Scholar] [CrossRef][Green Version]
- Dragoni, S.; Franco, G.; Regoli, M.; Bracciali, M.; Morandi, V.; Sgaragli, G.; Bertelli, E.; Valoti, M. Gold Nanoparticles Uptake and Cytotoxicity Assessed on Rat Liver Precision-Cut Slices. Toxicol. Sci. 2012, 128, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamadani, M.Y.I.; Alzahrani, A.M.; Yousef, M.I.; Kamel, M.A.; El-Sayed, W.M. Gold Nanoparticles Perturb Drug-Metabolizing Enzymes and Antioxidants in the Livers of Male Rats: Potential Impact on Drug Interactions. Int. J. Nanomed. 2020, 15, 5005–5016. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Qasim, M.; Park, C.; Yoo, H.; Kim, J.-H.; Hong, K. Cytotoxic Potential and Molecular Pathway Analysis of Silver Nanoparticles in Human Colon Cancer Cells HCT116. Int. J. Mol. Sci. 2018, 19, 2269. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wu, Y.; Zhang, J.; Wang, H.; Xie, X.; Ye, X.; Peng, D.; Chen, W. Induction of Cytochrome P450 Involved in the Accelerated Blood Clearance Phenomenon Induced by PEGylated Liposomes In Vivo. Drug Metab. Dispos. 2019, 47, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Korashy, H.M.; El-Kadi, A.O. Regulatory mechanisms modulating the expression of cytochrome P450 1A1 gene by heavy metals. Toxicol. Sci. 2005, 88, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Elbekai, R.H.; El-Kadi, A.O.S. Transcriptional activation and posttranscriptional modification of Cyp1a1 by arsenite, cadmium, and chromium. Toxicol. Lett. 2007, 172, 106–119. [Google Scholar] [CrossRef]
- Ashino, T.; Yamamoto, M.; Numazawa, S. Nrf2 Antioxidative System is Involved in Cytochrome P450 Gene Expression and Activity: A Delay in Pentobarbital Metabolism in Nrf2-Deficient Mice. Drug Metab. Dispos. 2020, 48, 673–680. [Google Scholar] [CrossRef]
- Abu-Bakar, A.; Satarug, S.; Marks, G.C.; Lang, M.A.; Moore, M.R. Acute cadmium chloride administration induces hepatic and renal CYP2A5 mRNA, protein and activity in the mouse: Involvement of transcription factor NRF2. Toxicol. Lett. 2004, 148, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.A.; Williams, T.D.; Chipman, J.K. Functional Analysis of a Metal Response Element in the Regulatory Region of Flounder Cytochrome P450 1A and Implications for Environmental Monitoring of Pollutants. Toxicol. Sci. 2006, 92, 387–393. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, H.; Baliga, R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003, 63, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhou, X.; Zhou, J.; Zhou, H.; Hong, X.; Li, D.; Xiang, Y.; Zhong, M.; Chen, Y.; Liang, D.; et al. Limonin mitigates cisplatin-induced acute kidney injury through metabolic reprogramming. Biomed. Pharmacother. 2023, 167, 115531. [Google Scholar] [CrossRef]
- Bakewell, S.; Conde, I.; Fallah, Y.; McCoy, M.; Jin, L.; Shajahan-Haq, A.N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers 2020, 12, 2647. [Google Scholar] [CrossRef] [PubMed]
- Baier, D.; Mendrina, T.; Schoenhacker-Alte, B.; Pirker, C.; Mohr, T.; Rusz, M.; Regner, B.; Schaier, M.; Sgarioto, N.; Raynal, N.J.-M.; et al. The Lipid Metabolism as Target and Modulator of BOLD-100 Anticancer Activity: Crosstalk with Histone Acetylation. Adv. Sci. 2023, 10, 2301939. [Google Scholar] [CrossRef] [PubMed]
- Petrunak, E.M.; Bart, A.G.; Peng, H.-M.; Auchus, R.J.; Scott, E.E. Human cytochrome P450 17A1 structures with metabolites of prostate cancer drug abiraterone reveal substrate-binding plasticity and a second binding site. J. Biol. Chem. 2023, 299, 102999. [Google Scholar] [CrossRef] [PubMed]
- Çekiç, S.Z.; Holtmann, D.; Güven, G.; Mangold, K.-M.; Schwaneberg, U.; Schrader, J. Mediated electron transfer with P450cin. Electrochem. Commun. 2010, 12, 1547–1550. [Google Scholar] [CrossRef]
- Panneerselvam, S.; Shehzad, A.; Mueller-Dieckmann, J.; Wilmanns, M.; Bocola, M.; Davari, M.D.; Schwaneberg, U. Crystallographic insights into a cobalt (III) sepulchrate based alternative cofactor system of P450 BM3 monooxygenase. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2018, 1866, 134–140. [Google Scholar] [CrossRef]
- Lazzara, P.R.; Moore, T.W. Scaffold-hopping as a strategy to address metabolic liabilities of aromatic compounds. RSC Med. Chem. 2020, 11, 18–29. [Google Scholar] [CrossRef]
- Dahal, U.P.; Joswig-Jones, C.; Jones, J.P. Comparative study of the affinity and metabolism of type I and type II binding quinoline carboxamide analogues by cytochrome P450 3A4. J. Med. Chem. 2012, 55, 280–290. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CYP | Assay | Protein Binding, Dark | Protein Binding, Light | Parent Ligand Protein Binding, | Dark Inhibition, | Light Inhibition, | Parent Ligand, |
|---|---|---|---|---|---|---|---|---|
| Kd (nM) | Kd (nM) | Kd (nM) | IC50 (µM) | IC50 (µM) | IC50 (µM) | |||
| 1 | 102A1 (P450BM3) | Protein | 6.8 | 0.05 | 0.06 | |||
| 2 | 17A1 | Protein | 89 | <100 | ||||
| 3 | 1B1 | Cells | 1.9 | 0.0003 | 0.00031 | |||
| 1A1 | Cells | >30 | >30 | 4.57 | ||||
| 19A1 | Cells | >30 | 18.8 | 14.4 | ||||
| phLM | Microsomes | >30 | 1.28 | 0.33 | ||||
| 4 | 3A4 | Protein | 340 | 0.9 | 2.2 | 1.54 | ||
| 5 | 3A4 | Protein | 70 | 0.25 | ||||
| 6 | 3A4 | Protein | 2.3 | |||||
| 3A4 | Cells | 16 | ||||||
| 1A2 | Protein | 72 | ||||||
| 2C9 | Protein | 17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havrylyuk, D.; Heidary, D.K.; Glazer, E.C. The Impact of Inorganic Systems and Photoactive Metal Compounds on Cytochrome P450 Enzymes and Metabolism: From Induction to Inhibition. Biomolecules 2024, 14, 441. https://doi.org/10.3390/biom14040441
Havrylyuk D, Heidary DK, Glazer EC. The Impact of Inorganic Systems and Photoactive Metal Compounds on Cytochrome P450 Enzymes and Metabolism: From Induction to Inhibition. Biomolecules. 2024; 14(4):441. https://doi.org/10.3390/biom14040441
Chicago/Turabian StyleHavrylyuk, Dmytro, David K. Heidary, and Edith C. Glazer. 2024. "The Impact of Inorganic Systems and Photoactive Metal Compounds on Cytochrome P450 Enzymes and Metabolism: From Induction to Inhibition" Biomolecules 14, no. 4: 441. https://doi.org/10.3390/biom14040441
APA StyleHavrylyuk, D., Heidary, D. K., & Glazer, E. C. (2024). The Impact of Inorganic Systems and Photoactive Metal Compounds on Cytochrome P450 Enzymes and Metabolism: From Induction to Inhibition. Biomolecules, 14(4), 441. https://doi.org/10.3390/biom14040441