
Transient Receptor Potential Canonical 5 (TRPC5): Regulation of Heart Rate and Protection against Pathological Cardiac Hypertrophy

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Animals
2.2. Radio Telemetry of Conscious Haemodynamics
2.3. Two-Dimensional Ultrasound Echocardiography
2.4. Haemodynamic Response to Vasoactive Agents
2.5. Langendorff Isolated Perfused Heart
2.6. Suprarenal Abdominal Aortic Banding (AAB) Model of Cardiac Hypertrophy
2.7. Histology
2.8. RT-qPCR
2.9. Immunoblotting
2.10. Statistical Analysis
3. Results
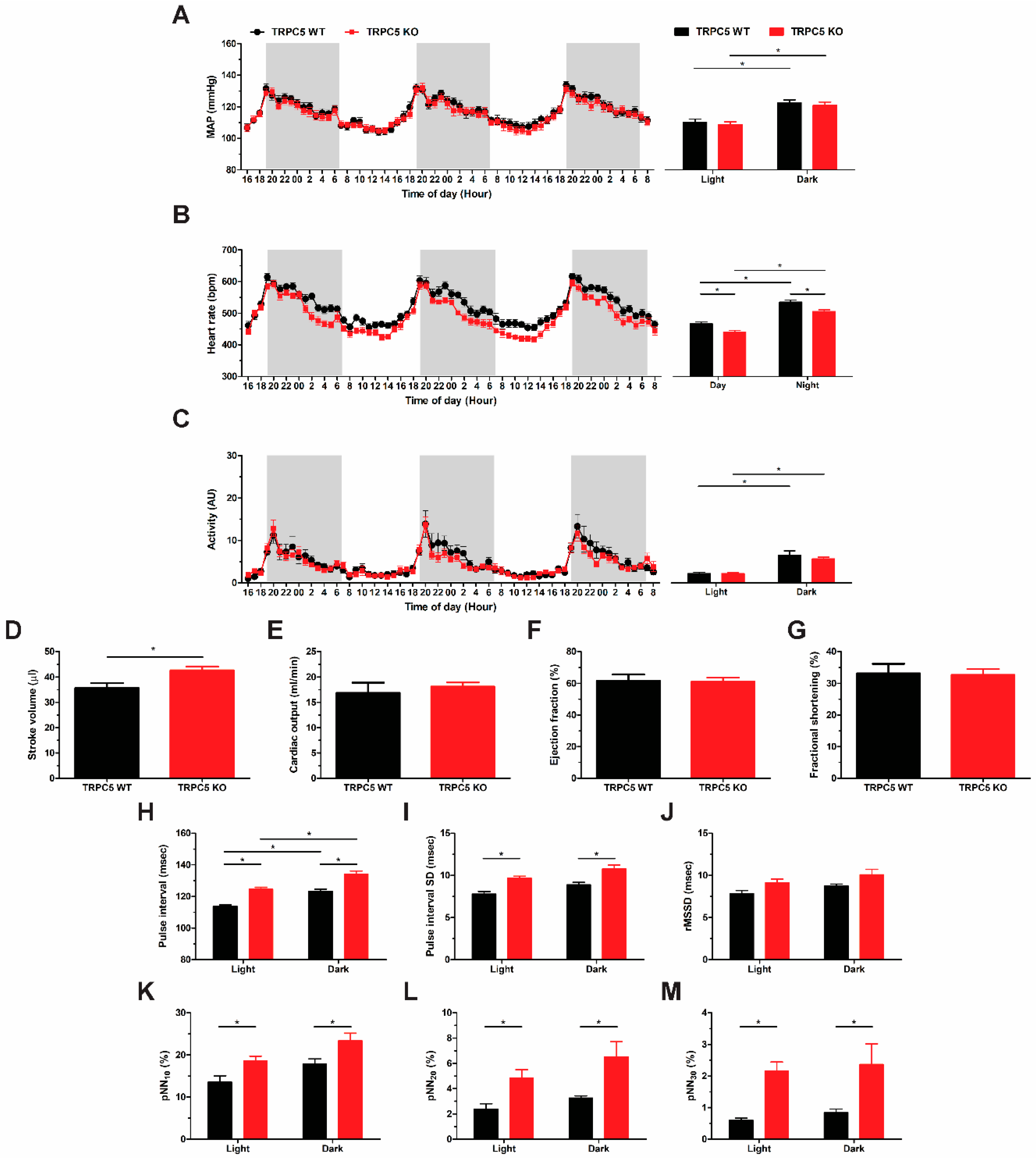
3.1. Baseline Reduction in Basal Heart Rate in Global TRPC5 KO Mice
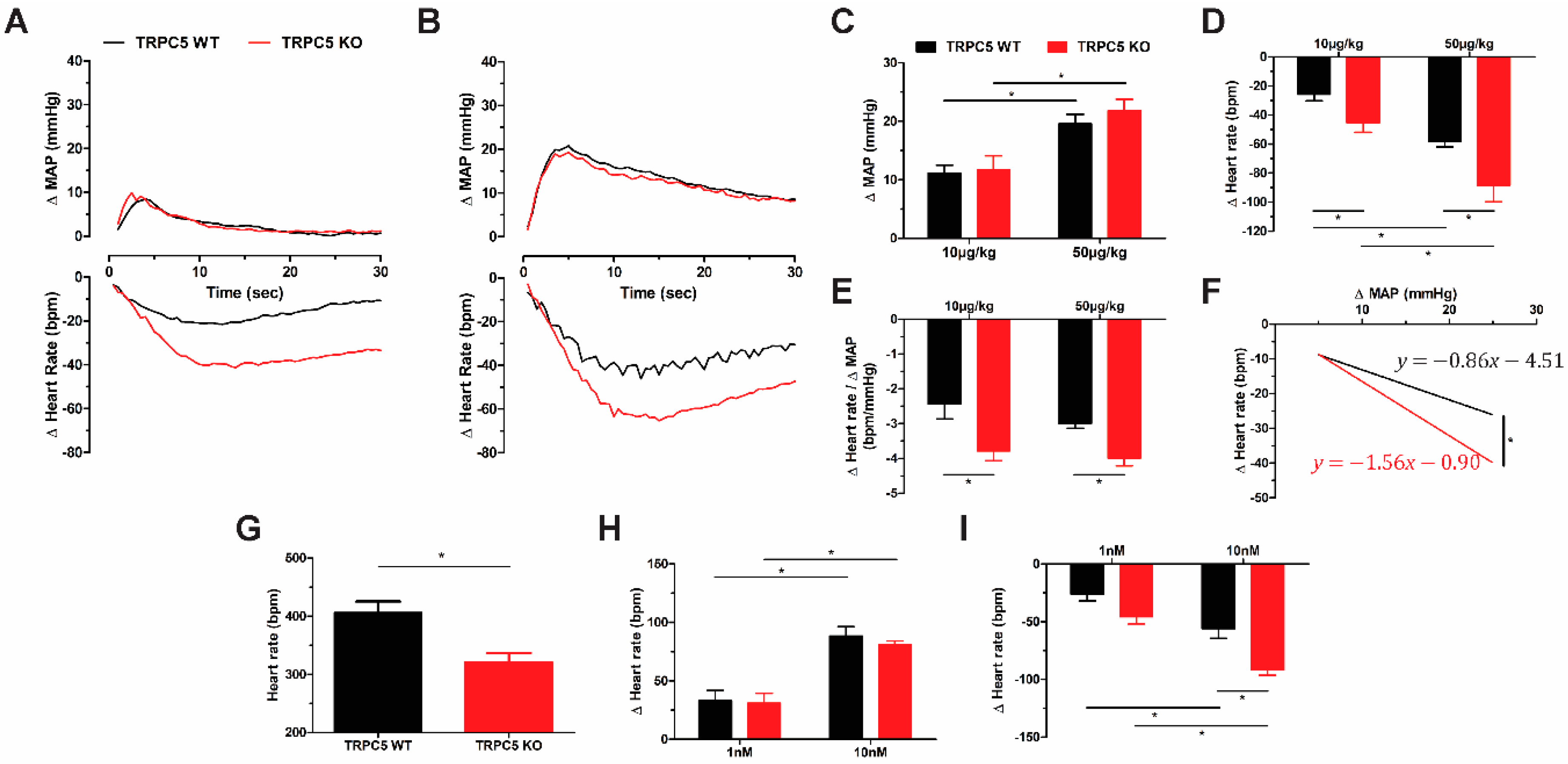
3.2. Cardiac Pacing Abnormalities and Enhanced Cardiac Response to Cholinergic Stimulation in TRPC5 KO Mice
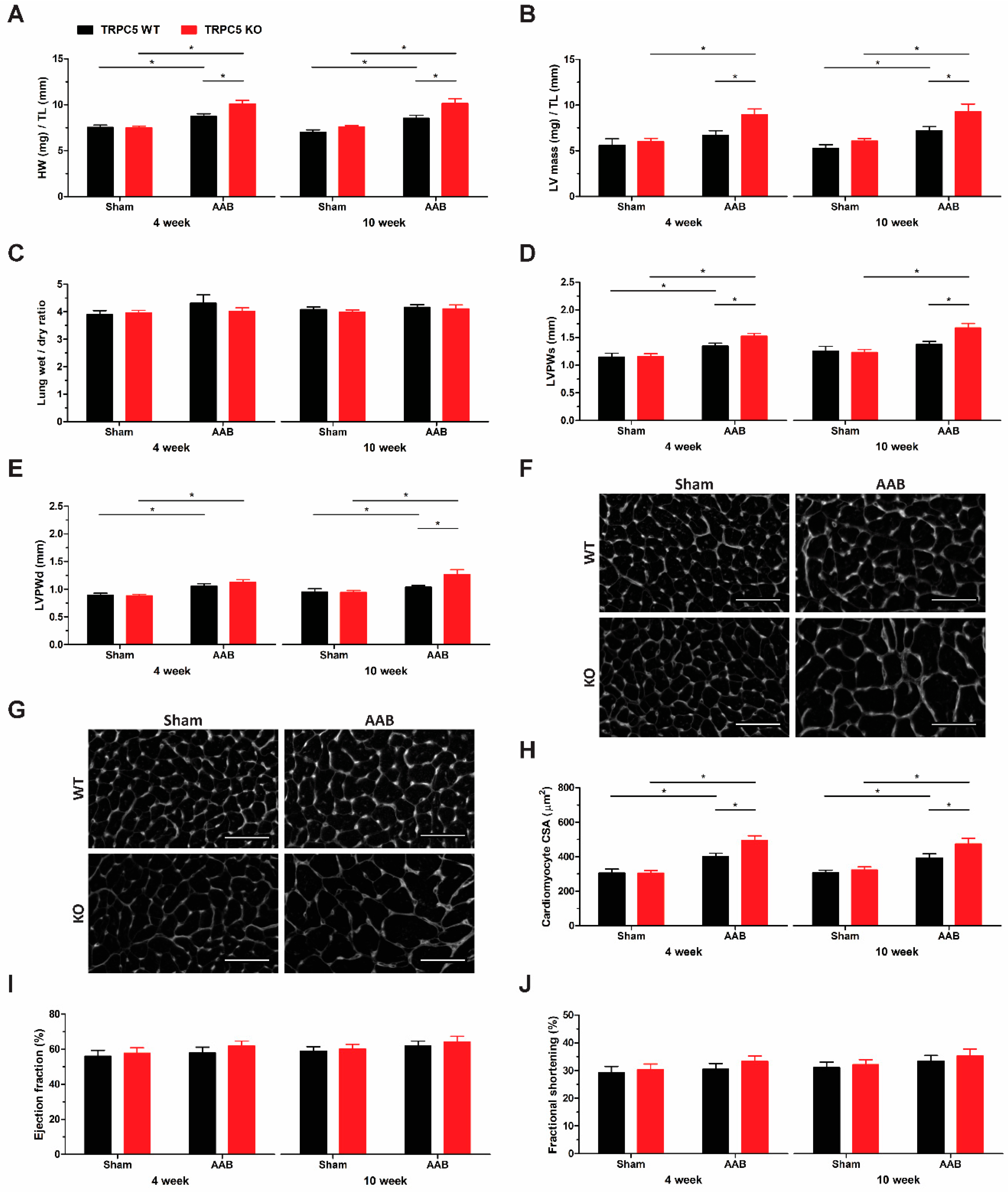
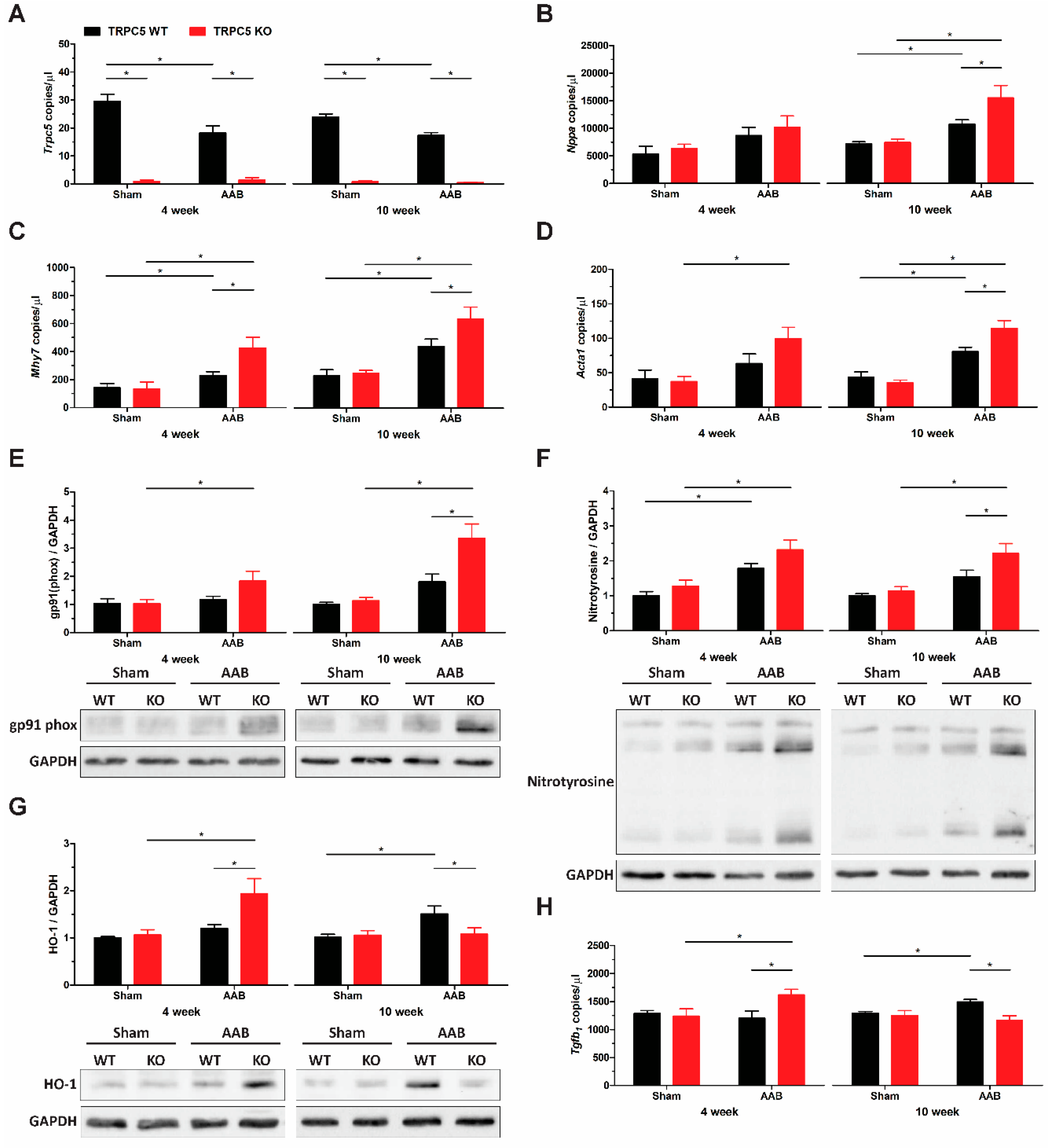
3.3. TRPC5 Protects against Pressure Overload-Induced Cardiac Hypertrophy
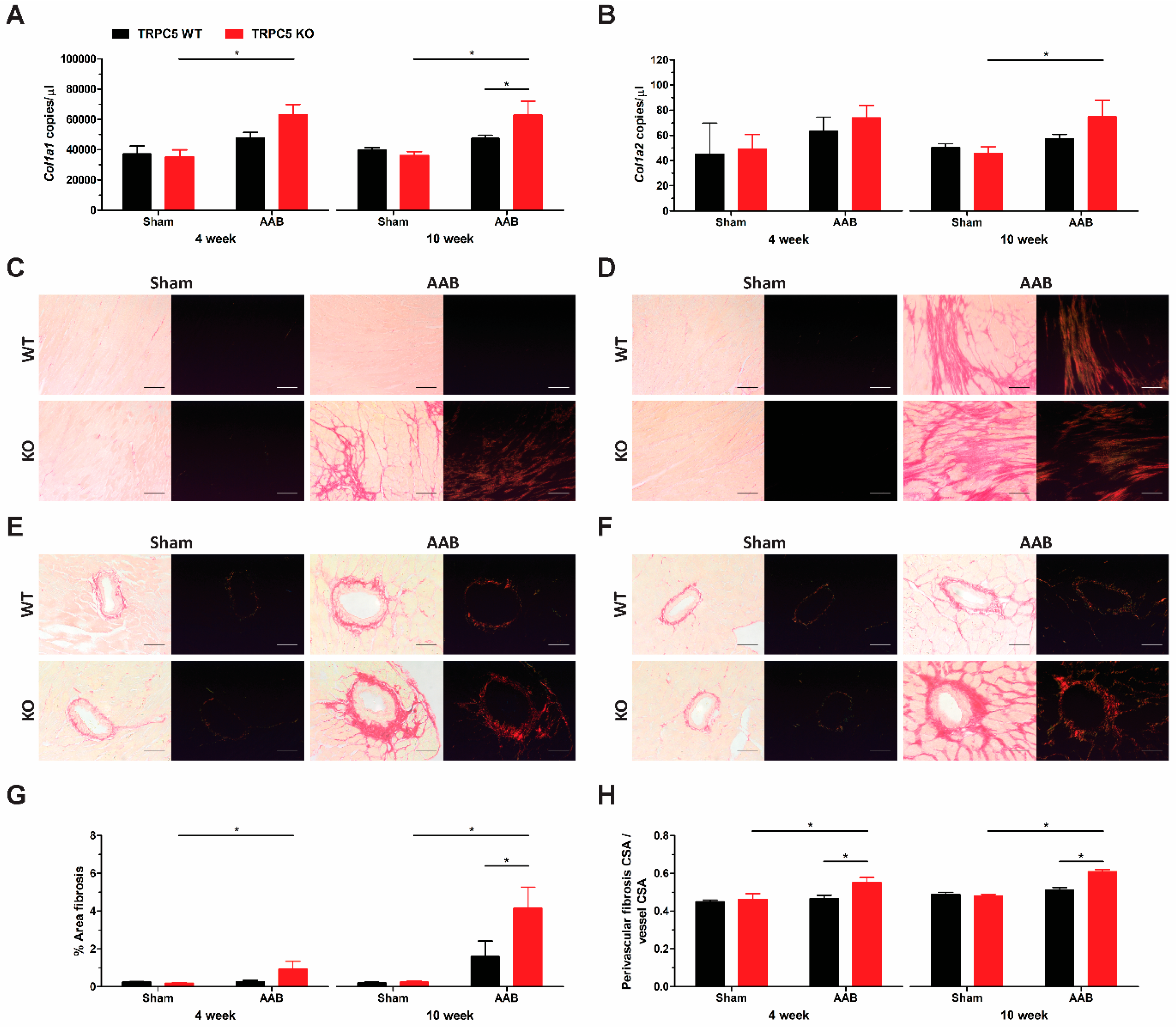
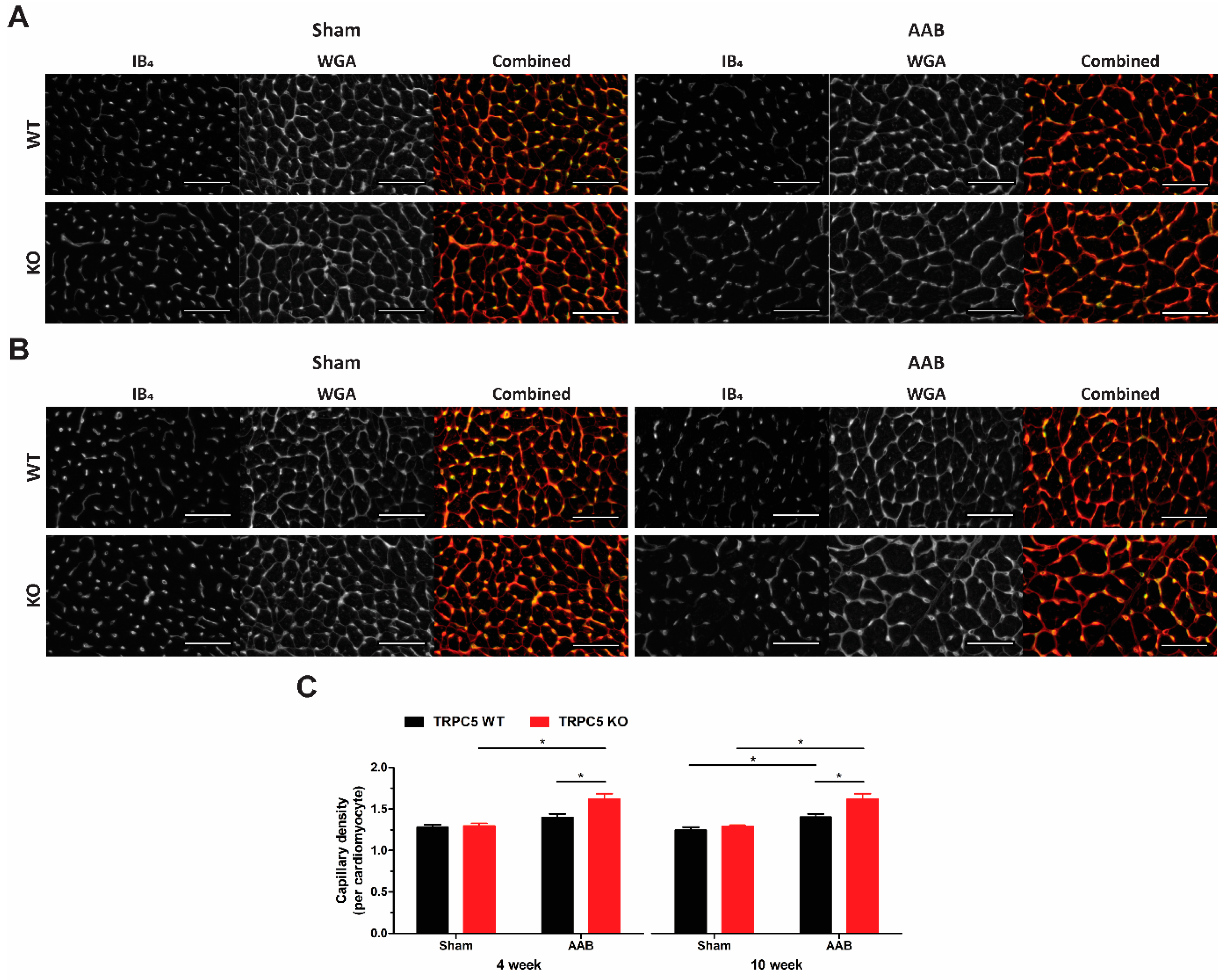
3.4. Exaggerated Hypertrophy Is Accompanied by Increased Left Ventricular Remodelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, re3. [Google Scholar] [CrossRef] [PubMed]
- Zholos, A.V. Trpc5. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2014; Volume 222, pp. 129–156. [Google Scholar] [CrossRef]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [CrossRef] [PubMed]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Medhurst, A.D.; Mattei, C.; Kelsell, R.E.; Calver, A.R.; Randall, A.D.; Benham, C.D.; Pangalos, M.N. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res. Mol. Brain Res. 2002, 109, 95–104. [Google Scholar] [CrossRef]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Boulay, G.; Flemming, R.; Beech, D.J. E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2653–H2659. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.N.; Albert, A.P.; Peppiatt-Wildman, C.M.; Large, W.A. Diverse properties of store-operated TRPC channels activated by protein kinase C in vascular myocytes. J. Physiol. 2008, 586, 2463–2476. [Google Scholar] [CrossRef]
- Bush, E.W.; Hood, D.B.; Papst, P.J.; Chapo, J.A.; Minobe, W.; Bristow, M.R.; Olson, E.N.; McKinsey, T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006, 281, 33487–33496. [Google Scholar] [CrossRef]
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507. [Google Scholar] [CrossRef]
- Saleh, S.N.; Albert, A.P.; Large, W.A. Activation of native TRPC1/C5/C6 channels by endothelin-1 is mediated by both PIP3 and PIP2 in rabbit coronary artery myocytes. J. Physiol. 2009, 587, 5361–5375. [Google Scholar] [CrossRef]
- Carson, C.; Raman, P.; Tullai, J.; Xu, L.; Henault, M.; Thomas, E.; Yeola, S.; Lao, J.; McPate, M.; Verkuyl, J.M.; et al. Englerin A Agonizes the TRPC4/C5 Cation Channels to Inhibit Tumor Cell Line Proliferation. PLoS ONE 2015, 10, e0127498. [Google Scholar] [CrossRef]
- Lau, O.C.; Shen, B.; Wong, C.O.; Tjong, Y.W.; Lo, C.Y.; Wang, H.C.; Huang, Y.; Yung, W.H.; Chen, Y.C.; Fung, M.L.; et al. TRPC5 channels participate in pressure-sensing in aortic baroreceptors. Nat. Commun. 2016, 7, 11947. [Google Scholar] [CrossRef]
- Thakore, P.; Brain, S.D.; Beech, D.J. Correspondence: Challenging a proposed role for TRPC5 in aortic baroreceptor pressure-sensing. Nat. Commun. 2018, 9, 1245. [Google Scholar] [CrossRef]
- Lau, O.C.; Shen, B.; Wong, C.O.; Yao, X. Correspondence: Reply to ‘Challenging a proposed role for TRPC5 in aortic baroreceptor pressure-sensing’. Nat. Commun. 2018, 9, 1244. [Google Scholar] [CrossRef]
- Sabourin, J.; Robin, E.; Raddatz, E. A key role of TRPC channels in the regulation of electromechanical activity of the developing heart. Cardiovasc. Res. 2011, 92, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.K.; Chu, Y.; Chaulet, H.; Lai, D.; Gervasio, O.L.; Graham, R.M.; Cannell, M.B.; Allen, D.G. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ. Res. 2007, 100, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Seth, M.; Sumbilla, C.; Mullen, S.P.; Lewis, D.; Klein, M.G.; Hussain, A.; Soboloff, J.; Gill, D.L.; Inesi, G. Sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) gene silencing and remodeling of the Ca2+ signaling mechanism in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 16683–16688. [Google Scholar] [CrossRef]
- Liu, F.F.; Ma, Z.Y.; Li, D.L.; Feng, J.B.; Zhang, K.; Wang, R.; Zhang, W.; Li, L.; Zhang, Y. Differential expression of TRPC channels in the left ventricle of spontaneously hypertensive rats. Mol. Biol. Rep. 2010, 37, 2645–2651. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Wilkin, B.J.; Bodi, I.; Molkentin, J.D. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006, 20, 1660–1670. [Google Scholar] [CrossRef]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef]
- Sunggip, C.; Shimoda, K.; Oda, S.; Tanaka, T.; Nishiyama, K.; Mangmool, S.; Nishimura, A.; Numaga-Tomita, T.; Nishida, M. TRPC5-eNOS Axis Negatively Regulates ATP-Induced Cardiomyocyte Hypertrophy. Front. Pharmacol. 2018, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Li, Y.; Moon, J.; Kim, K.S.; Smith, K.S.; Rudolph, U.; Gapon, S.; Yao, G.L.; Tsvetkov, E.; Rodig, S.J.; et al. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell 2009, 137, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Aubdool, A.A.; Thakore, P.; Argunhan, F.; Smillie, S.J.; Schnelle, M.; Srivastava, S.; Alawi, K.M.; Wilde, E.; Mitchell, J.; Farrell-Dillon, K.; et al. A Novel alpha-Calcitonin Gene-Related Peptide Analogue Protects Against End-Organ Damage in Experimental Hypertension, Cardiac Hypertrophy and Heart Failure. Circulation 2017, 136, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Thireau, J.; Zhang, B.L.; Poisson, D.; Babuty, D. Heart rate variability in mice: A theoretical and practical guide. Exp. Physiol. 2008, 93, 83–94. [Google Scholar] [CrossRef]
- Sgoifo, A.; Carnevali, L.; Alfonso Mde, L.; Amore, M. Autonomic dysfunction and heart rate variability in depression. Stress 2015, 18, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Kassiri, Z.; Virag, J.A.I.; de Castro Bras, L.E.; Scherrer-Crosbie, M. Guidelines for measuring cardiac physiology in mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H733–H752. [Google Scholar] [CrossRef] [PubMed]
- Pamidimukkala, J.; Xue, B.; Newton, L.G.; Lubahn, D.B.; Hay, M. Estrogen receptor-alpha mediates estrogen facilitation of baroreflex heart rate responses in conscious mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1063–H1070. [Google Scholar] [CrossRef]
- Gu, H.; Lin, M.; Liu, J.; Gozal, D.; Scrogin, K.E.; Wurster, R.; Chapleau, M.W.; Ma, X.; Cheng, Z.J. Selective impairment of central mediation of baroreflex in anesthetized young adult Fischer 344 rats after chronic intermittent hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2809–H2818. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Stein, P.K.; Bosner, M.S.; Kleiger, R.E.; Conger, B.M. Heart rate variability: A measure of cardiac autonomic tone. Am. Heart J. 1994, 127, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Laude, D.; Baudrie, V.; Elghozi, J.L. Effects of atropine on the time and frequency domain estimates of blood pressure and heart rate variability in mice. Clin. Exp. Pharmacol. Physiol. 2008, 35, 454–457. [Google Scholar] [CrossRef]
- Yamada, M.; Inanobe, A.; Kurachi, Y. G protein regulation of potassium ion channels. Pharmacol. Rev. 1998, 50, 723–760. [Google Scholar] [PubMed]
- Brodde, O.E.; Michel, M.C. Adrenergic and muscarinic receptors in the human heart. Pharmacol. Rev. 1999, 51, 651–690. [Google Scholar] [PubMed]
- Jeon, J.P.; Hong, C.; Park, E.J.; Jeon, J.H.; Cho, N.H.; Kim, I.G.; Choe, H.; Muallem, S.; Kim, H.J.; So, I. Selective Galphai subunits as novel direct activators of transient receptor potential canonical (TRPC)4 and TRPC5 channels. J. Biol. Chem. 2012, 287, 17029–17039. [Google Scholar] [CrossRef]
- Barrick, C.J.; Rojas, M.; Schoonhoven, R.; Smyth, S.S.; Threadgill, D.W. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: Temporal- and background-dependent development of concentric left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2119–H2130. [Google Scholar] [CrossRef] [PubMed]
- Mito, S.; Ozono, R.; Oshima, T.; Yano, Y.; Watari, Y.; Yamamoto, Y.; Brydun, A.; Igarashi, K.; Yoshizumi, M. Myocardial protection against pressure overload in mice lacking Bach1, a transcriptional repressor of heme oxygenase-1. Hypertension 2008, 51, 1570–1577. [Google Scholar] [CrossRef]
- Hu, C.M.; Chen, Y.H.; Chiang, M.T.; Chau, L.Y. Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation 2004, 110, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huo, R.; Tong, Y.; Sheng, Y.; Liu, H.B.; Gao, X.; Nakajima, O.; Yang, B.F.; Dong, D.L. Systemic heme oxygenase-1 transgenic overexpression aggravates pressure overload-induced cardiac hypertrophy in mice. Cell. Physiol. Biochem. 2011, 28, 25–32. [Google Scholar] [CrossRef]
- Allwood, M.A.; Kinobe, R.T.; Ballantyne, L.; Romanova, N.; Melo, L.G.; Ward, C.A.; Brunt, K.R.; Simpson, J.A. Heme oxygenase-1 overexpression exacerbates heart failure with aging and pressure overload but is protective against isoproterenol-induced cardiomyopathy in mice. Cardiovasc. Pathol. 2014, 23, 231–237. [Google Scholar] [CrossRef]
- Ning, W.; Song, R.; Li, C.; Park, E.; Mohsenin, A.; Choi, A.M.; Choi, M.E. TGF-beta1 stimulates HO-1 via the p38 mitogen-activated protein kinase in A549 pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L1094–L1102. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Chau, L.Y. Heme oxygenase-1 in cardiovascular diseases: Molecular mechanisms and clinical perspectives. Chang Gung Med. J. 2010, 33, 13–24. [Google Scholar]
- Li, J.M.; Brooks, G. Differential protein expression and subcellular distribution of TGFbeta1, beta2 and beta3 in cardiomyocytes during pressure overload-induced hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2213–2224. [Google Scholar] [CrossRef]
- Yoshida, T.; Inoue, R.; Morii, T.; Takahashi, N.; Yamamoto, S.; Hara, Y.; Tominaga, M.; Shimizu, S.; Sato, Y.; Mori, Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006, 2, 596–607. [Google Scholar] [CrossRef]
- Rose, R.A.; Hatano, N.; Ohya, S.; Imaizumi, Y.; Giles, W.R. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J. Physiol. 2007, 580, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Jacobo, S.M.; Billing, D.; Rozkalne, A.; Gage, S.D.; Anagnostou, T.; Pavenstadt, H.; Hsu, H.H.; Schlondorff, J.; Ramos, A.; et al. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci. Signal. 2010, 3, ra77. [Google Scholar] [CrossRef] [PubMed]
- Tirziu, D.; Chorianopoulos, E.; Moodie, K.L.; Palac, R.T.; Zhuang, Z.W.; Tjwa, M.; Roncal, C.; Eriksson, U.; Fu, Q.; Elfenbein, A.; et al. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin. Investig. 2007, 117, 3188–3197. [Google Scholar] [CrossRef]
- Jaba, I.M.; Zhuang, Z.W.; Li, N.; Jiang, Y.; Martin, K.A.; Sinusas, A.J.; Papademetris, X.; Simons, M.; Sessa, W.C.; Young, L.H.; et al. NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J. Clin. Investig. 2013, 123, 1718–1731. [Google Scholar] [CrossRef]
- Accornero, F.; van Berlo, J.H.; Benard, M.J.; Lorenz, J.N.; Carmeliet, P.; Molkentin, J.D. Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ. Res. 2011, 109, 272–280. [Google Scholar] [CrossRef]
- Alawi, K.M.; Russell, F.A.; Aubdool, A.A.; Srivastava, S.; Riffo-Vasquez, Y.; Baldissera, L., Jr.; Thakore, P.; Saleque, N.; Fernandes, E.S.; Walsh, D.A.; et al. Transient receptor potential canonical 5 (TRPC5) protects against pain and vascular inflammation in arthritis and joint inflammation. Ann. Rheum. Dis. 2017, 76, 252–260. [Google Scholar] [CrossRef]
- Wollert, K.C.; Drexler, H. Regulation of cardiac remodeling by nitric oxide: Focus on cardiac myocyte hypertrophy and apoptosis. Heart Fail. Rev. 2002, 7, 317–325. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakore, P.; Clark, J.E.; Aubdool, A.A.; Thapa, D.; Starr, A.; Fraser, P.A.; Farrell-Dillon, K.; Fernandes, E.S.; McFadzean, I.; Brain, S.D. Transient Receptor Potential Canonical 5 (TRPC5): Regulation of Heart Rate and Protection against Pathological Cardiac Hypertrophy. Biomolecules 2024, 14, 442. https://doi.org/10.3390/biom14040442
Thakore P, Clark JE, Aubdool AA, Thapa D, Starr A, Fraser PA, Farrell-Dillon K, Fernandes ES, McFadzean I, Brain SD. Transient Receptor Potential Canonical 5 (TRPC5): Regulation of Heart Rate and Protection against Pathological Cardiac Hypertrophy. Biomolecules. 2024; 14(4):442. https://doi.org/10.3390/biom14040442
Chicago/Turabian StyleThakore, Pratish, James E. Clark, Aisah A. Aubdool, Dibesh Thapa, Anna Starr, Paul A. Fraser, Keith Farrell-Dillon, Elizabeth S. Fernandes, Ian McFadzean, and Susan D. Brain. 2024. "Transient Receptor Potential Canonical 5 (TRPC5): Regulation of Heart Rate and Protection against Pathological Cardiac Hypertrophy" Biomolecules 14, no. 4: 442. https://doi.org/10.3390/biom14040442