
Enhancement of Acetate-Induced Apoptosis of Colorectal Cancer Cells by Cathepsin D Inhibition Depends on Oligomycin A-Sensitive Respiration
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Sulforhodamine (SRB) Assay
2.3. Determination of O2 Consumption
2.4. Caspase-3 Activity
2.5. Flow Cytometry
2.6. Statistical Analysis
3. Results
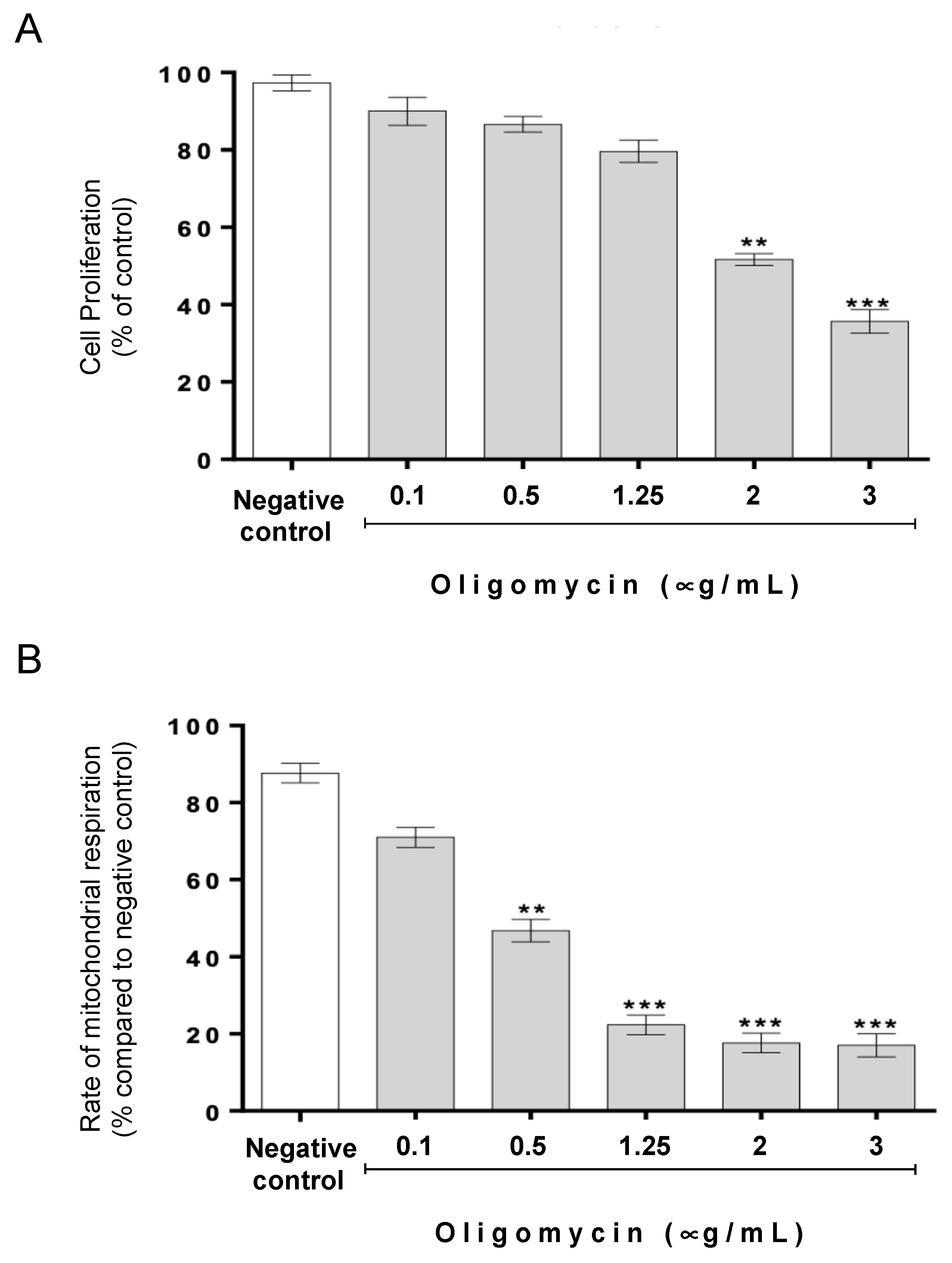
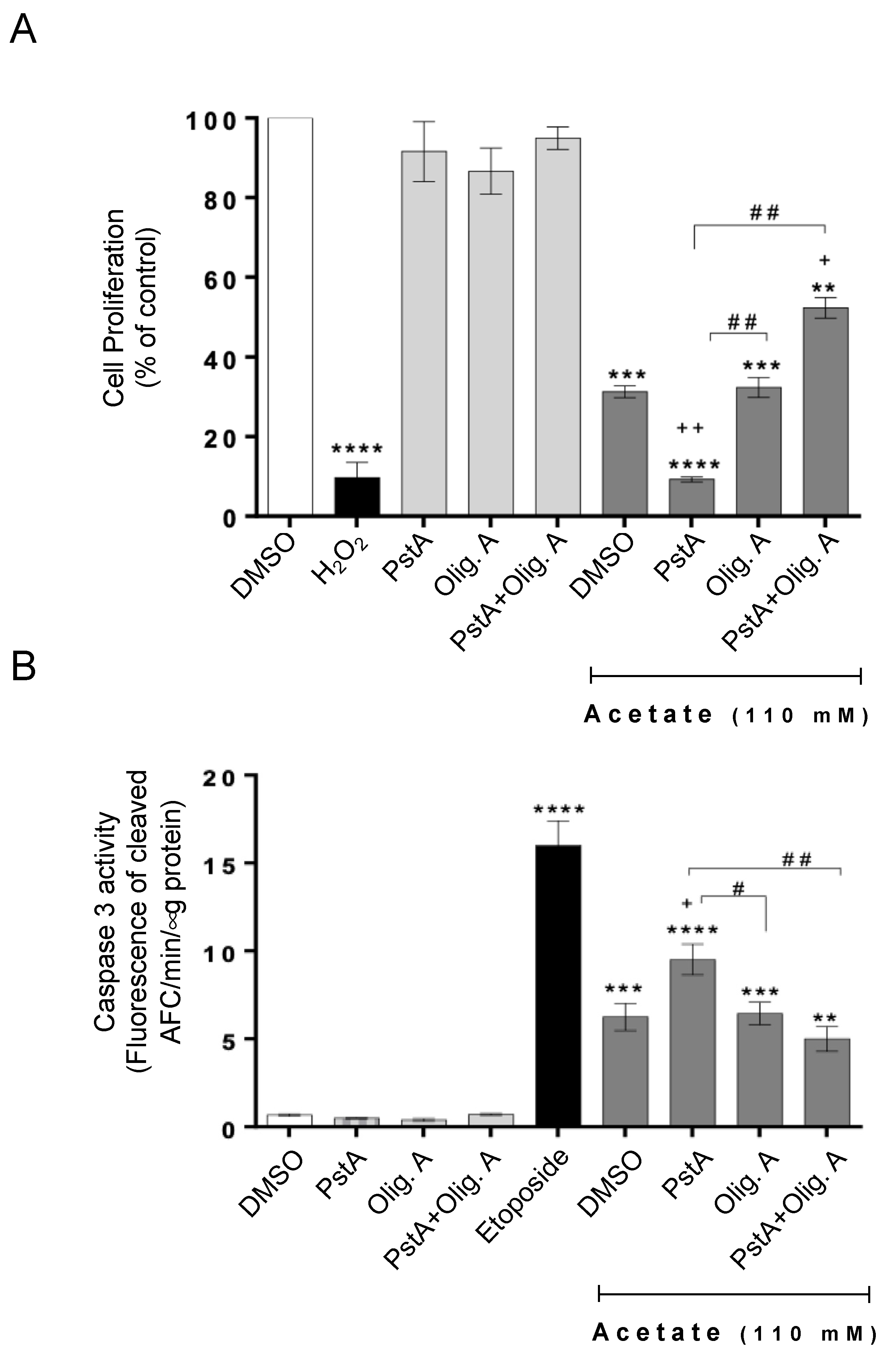
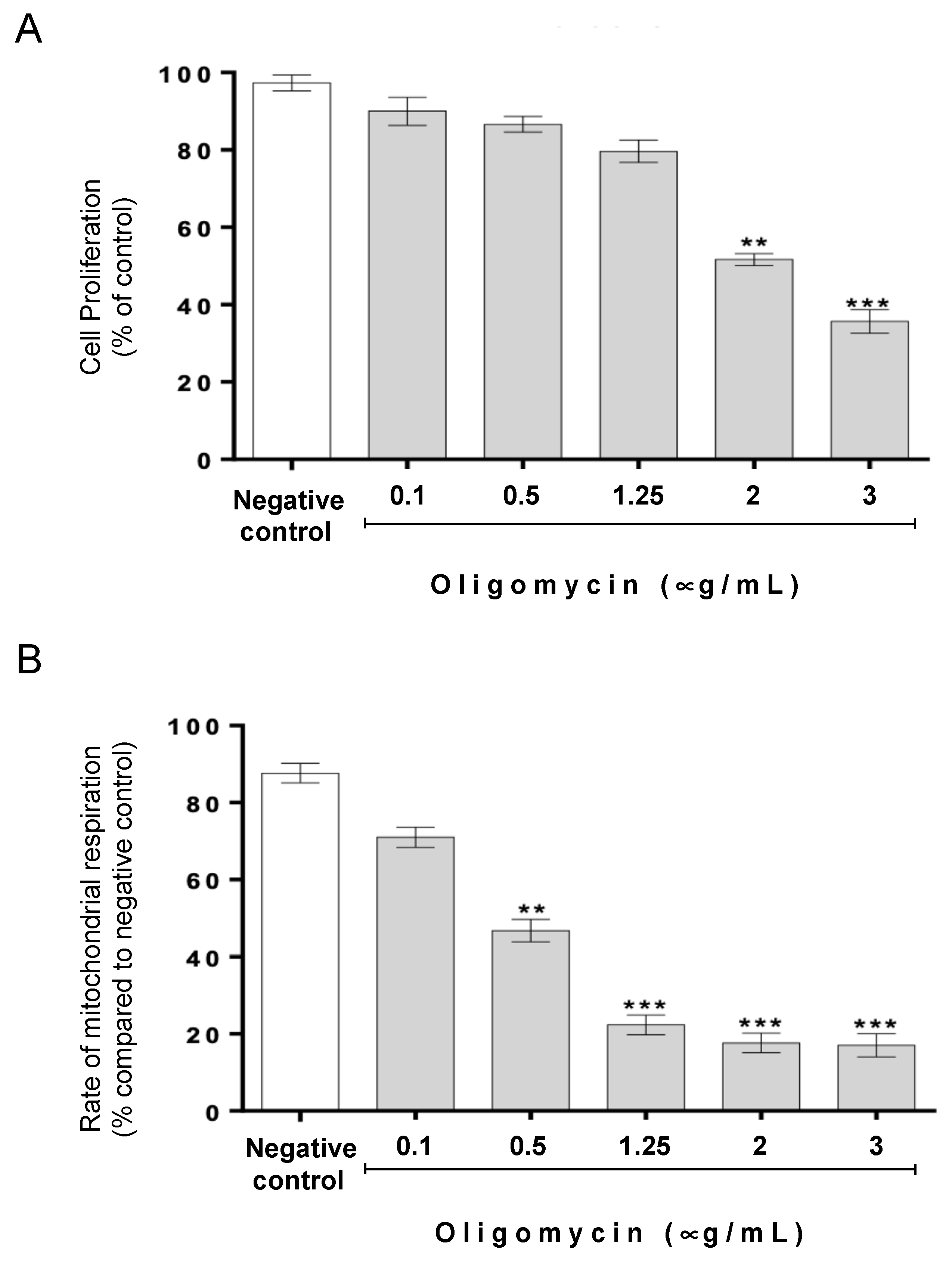
3.1. The Protective Role of Cathepsin D in Acetate-Induced Apoptosis Depends on Oligomycin A-Sensitive Respiration
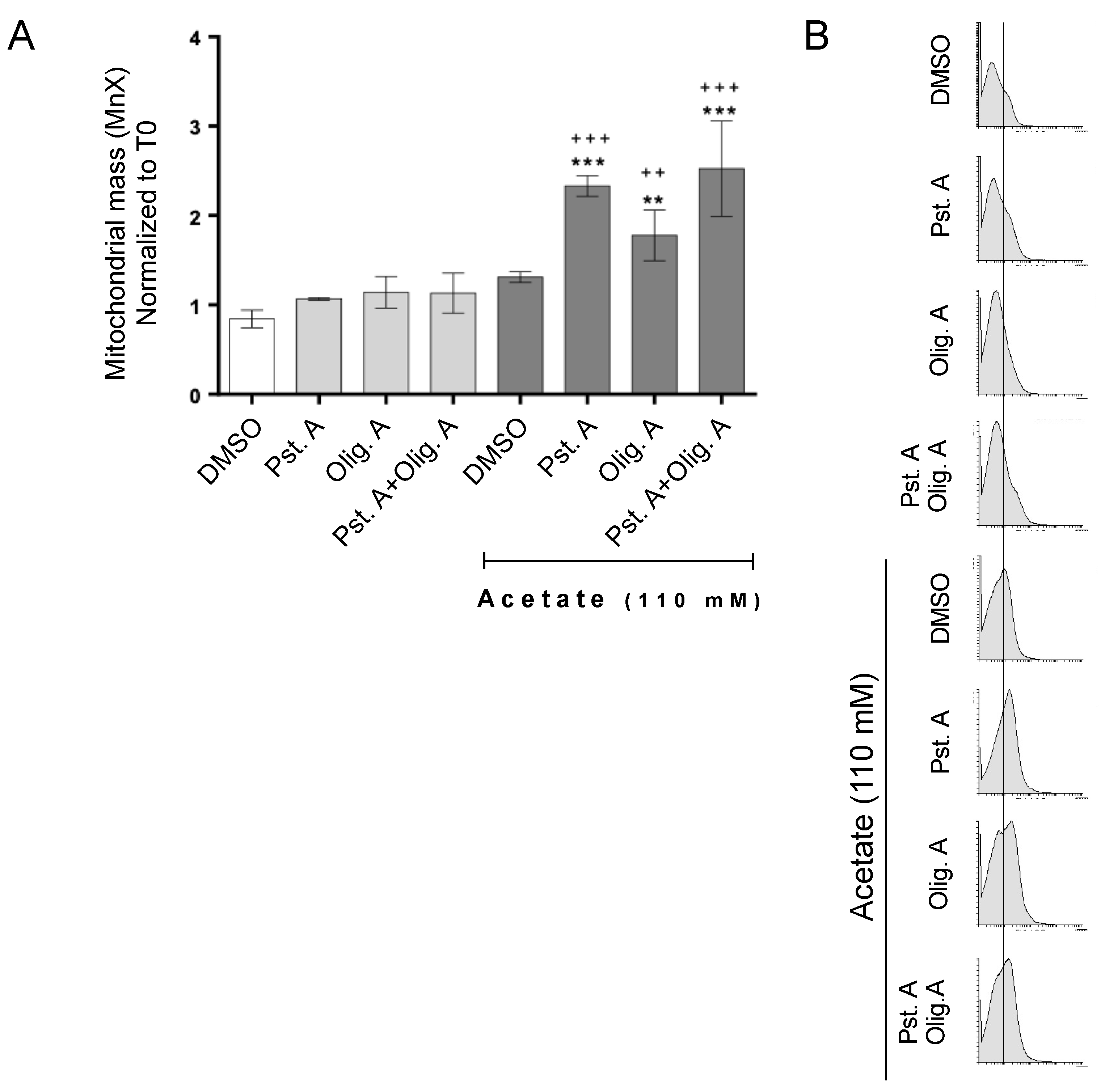
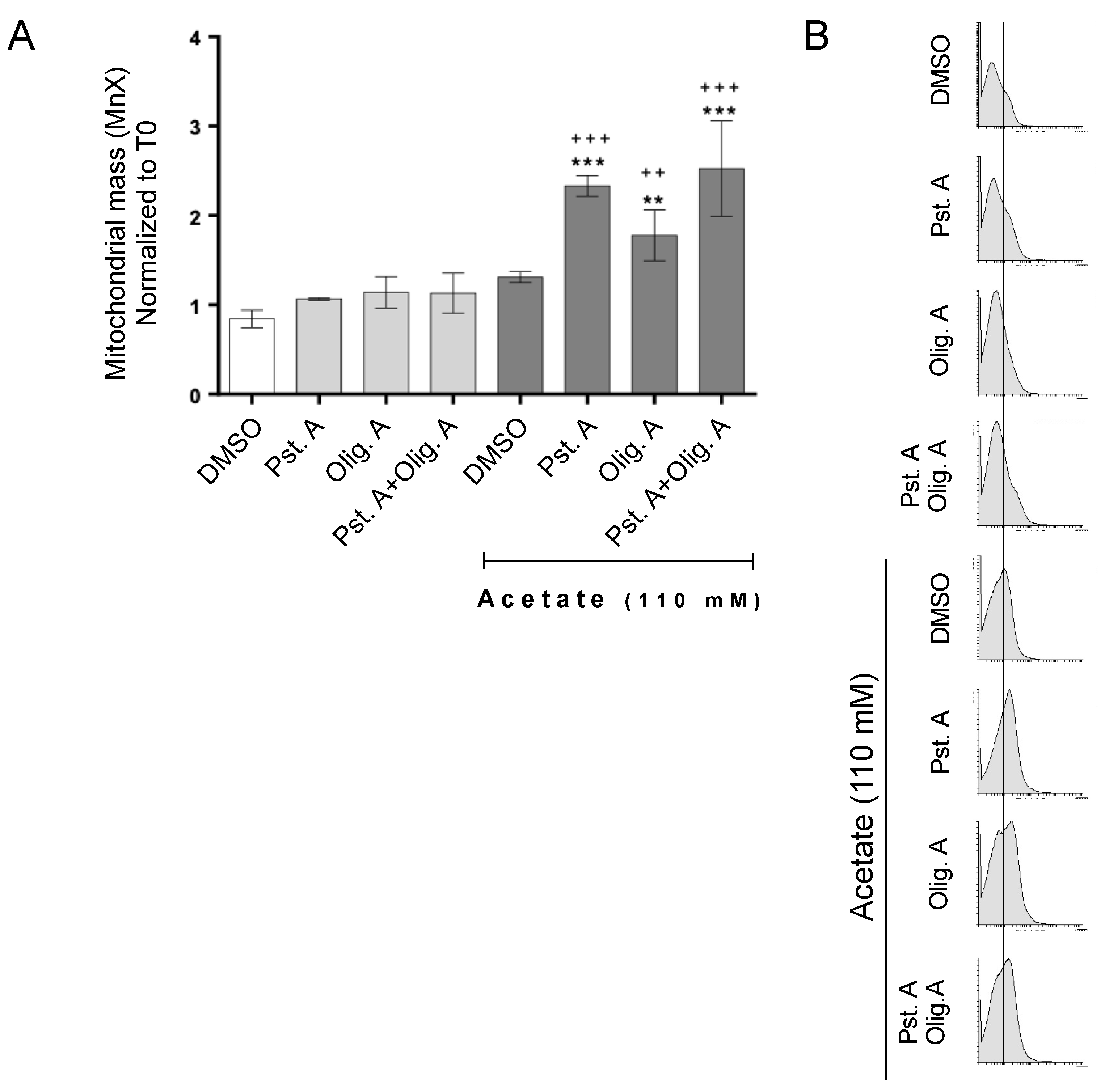
3.2. The Role of Cathepsin D in Mitochondrial Degradation Is Not Affected by Oligomycin A-Sensitive Respiration
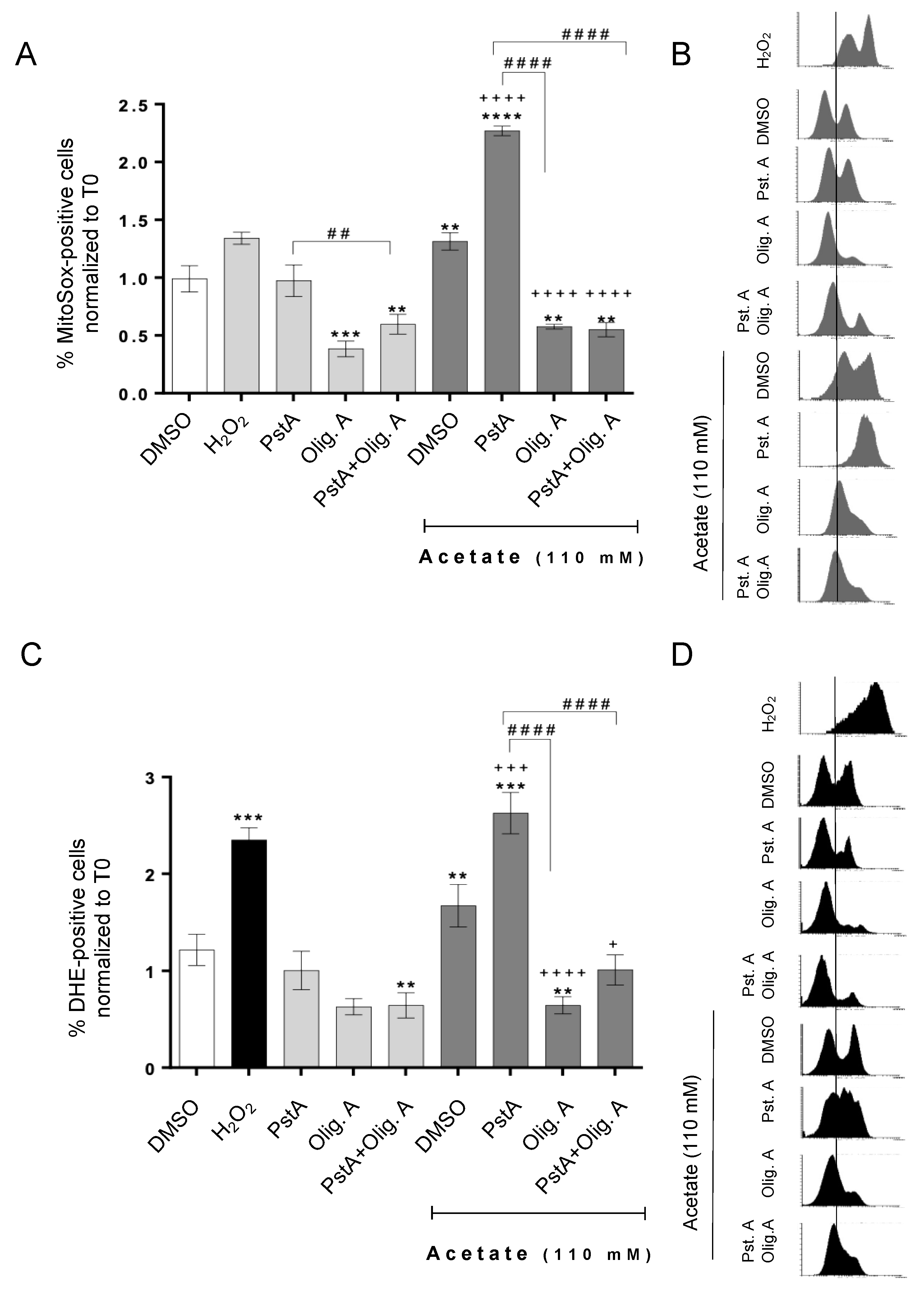
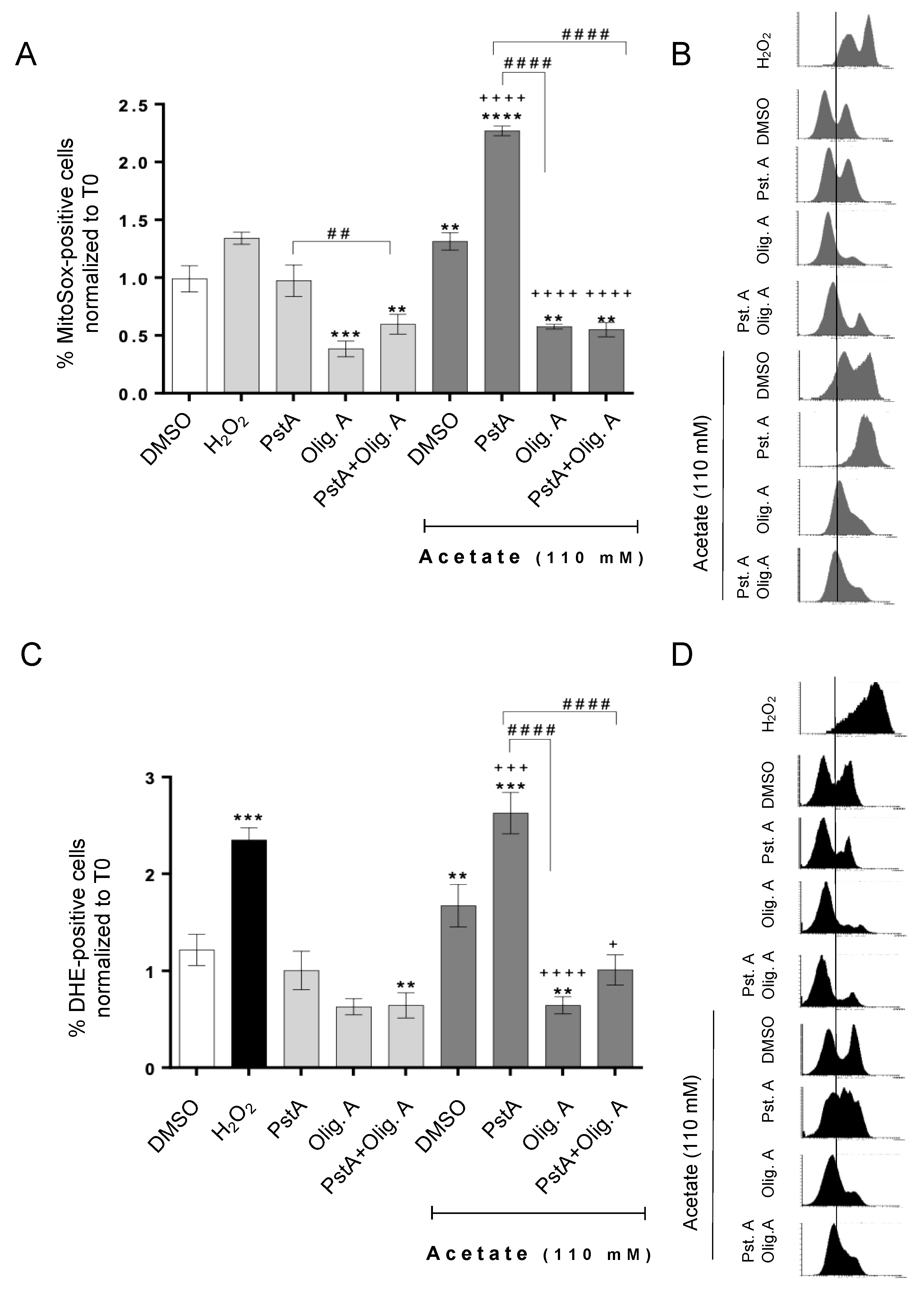
3.3. Oligomycin A Decreases ROS Accumulation Induced by Acetate Independently of Cathepsin D Activity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | colorectal cancer; |
| CatD | cathepsin D; |
| DHE | dihydroethidium; |
| OligA | oligomycin A; |
| PstA | pepstatin A; |
| ROS | reactive oxygen species; |
| RT | room temperature |
| SCFA | short-chain fatty acids; |
| SRB | sulforhodamine B |
References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Blondy, S.; David, V.; Verdier, M.; Mathonnet, M.; Perraud, A.; Christou, N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Baltazar, F.; Silva, E.; Preto, A. Microbiota-Derived Short-Chain Fatty Acids: New Road in Colorectal Cancer Therapy. Pharmaceutics 2022, 14, 2359. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.K. Potential Role of the Gut Microbiome In Colorectal Cancer Progression. Front. Immunol. 2022, 12, 807648. [Google Scholar] [CrossRef] [PubMed]
- Alvandi, E.; Wong, W.K.M.; Joglekar, M.V.; Spring, K.J.; Hardikar, A.A. Short-chain fatty acid concentrations in the incidence and risk-stratification of colorectal cancer: A systematic review and meta-analysis. BMC Med. 2022, 20, 323. [Google Scholar] [CrossRef] [PubMed]
- Peluzio, M.D.C.G.; Martinez, J.A.; Milagro, F.I. Postbiotics: Metabolites and mechanisms involved in microbiota-host interactions. Trends Food Sci. Technol. 2021, 108, 11–26. [Google Scholar] [CrossRef]
- Fotiadis, C.I.; Stoidis, C.N.; Spyropoulos, B.G.; Zografos, E.D. Role of probiotics, prebiotics and synbiotics in chemoprevention for colorectal cancer. World J. Gastroenterol. 2008, 14, 6453–6457. [Google Scholar] [CrossRef] [PubMed]
- Jan, G.; Belzacq, A.-S.; Haouzi, D.; Rouault, A.; Métivier, D.; Kroemer, G.; Brenner, C. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002, 9, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Bruneau, A.; Bensaada, M.; Philippe, C.; Bellaud, P.; Rabot, S.; Jan, G. Increased induction of apoptosis by Propionibacterium freudenreichii TL133 in colonic mucosal crypts of human microbiota-associated rats treated with 1,2-dimethylhydrazine. Br. J. Nutr. 2008, 100, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Comalada, M.; Bailón, E.; de Haro, O.; Lara-Villoslada, F.; Xaus, J.; Zarzuelo, A.; Gálvez, J. The effects of short-chain fatty acids on colon epithelial proliferation and survival depend on the cellular phenotype. J. Cancer Res. Clin. Oncol. 2006, 132, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Scheppach, W.; Bartram, H.; Richter, F. Role of short-chain fatty acids in the prevention of colorectal cancer. Eur. J. Cancer 1995, 31, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Mowat, C.; Dhatt, J.; Bhatti, I.; Hamie, A.; Baker, K. Short chain fatty acids prime colorectal cancer cells to activate antitumor immunity. Front. Immunol. 2023, 14, 1190810. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.S.F.; Pereira, H.; Alves, S.; Castro, L.; Baltazar, F.; Chaves, S.R.; Preto, A.; Côrte-Real, M. Cathepsin D protects colorectal cancer cells from acetate-induced apoptosis through autophagyindependent degradation of damaged mitochondria. Cell Death Dis. 2015, 6, e1788. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Oliveira, C.S.F.; Alves, S.; Chaves, S.R.; Coutinho, O.P.; Côrte-Real, M.; Preto, A. Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin D release. Cell Death Dis. 2013, 4, e507. [Google Scholar] [CrossRef] [PubMed]
- Ferro, S.; Azevedo-Silva, J.; Casal, M.; Côrte-Real, M.; Baltazar, F.; Preto, A. Characterization of acetate transport in colorectal cancer cells and potential therapeutic implications. Oncotarget 2016, 7, 70639–70653. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.Y.; Sung, N.Y.; Lee, Y.S.; Kwon, T.S.; Si, Y.; Lee, Y.S.; Oh, S.T.; Lee, I.K. The expression of multiple proteins as prognostic factors in colorectal cancer: Cathepsin D, p53, COX-2, epidermal growth factor receptor, C-erbB-2, and Ki-67. Gut Liver 2014, 8, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Sebzda, T.; Saleh, Y.; Gburek, J.; Andrzejak, R.; Gnus, J.; Siewinski, M.; Grzebieniak, Z. Cathepsin D expression in human colorectal cancer: Relationship with tumour type and tissue differentiation grade. J. Exp. Ther. Oncol. 2005, 5, 145–150. [Google Scholar] [PubMed]
- Pereira, C.; Chaves, S.; Alves, S.; Salin, B.; Camougrand, N.; Manon, S.; Sousa, M.J.; Côrte-Real, M. Mitochondrial degradation in acetic acid-induced yeast apoptosis: The role of Pep4 and the ADP/ATP carrier. Mol. Microbiol. 2010, 76, 1398–1410. [Google Scholar] [CrossRef]
- Pereira, H.P. The Role of Pep4p, the Vacuolar Yeast Protease Ortholog of Human Cathepsin D, in Mitochondria-Dependent Apoptosis. Ph.D. Thesis, Universidade do Minho, Braga, Portugal, 2015. Available online: https://hdl.handle.net/1822/38434 (accessed on 1 April 2024).
- Ocampo, A.; Liu, J.; Schroeder, E.A.; Shadel, G.S.; Barrientos, A. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 2012, 16, 55–67. [Google Scholar] [CrossRef]
- Devin, A.; Dejean, L.; Beauvoit, B.; Chevtzoff, C.; Avéret, N.; Bunoust, O.; Rigoulet, M. Growth yield homeostasis in respiring yeast is due to a strict mitochondrial content adjustment. J. Biol. Chem. 2006, 281, 26779–26784. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Giorgio, V.; Fogolari, F.; Glick, G.; Bernardi, P.; Lippe, G. The Oligomycin-Sensitivity Conferring Protein of Mitochondrial ATP Synthase: Emerging New Roles in Mitochondrial Pathophysiology. Int. J. Mol. Sci. 2014, 15, 7513–7536. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Radpour, R. New trends in molecular and cellular biomarker discovery for colorectal cancer. World J. Gastroenterol. 2016, 22, 5678–5693. [Google Scholar] [CrossRef] [PubMed]
- Palma, S.; Zwenger, A.O.; Croce, M.V.; Abba, M.C.; Lacunza, E. From molecular biology to clinical trials: Toward personalized colorectal cancer therapy. Clin. Color. Cancer 2016, 15, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Lipsyc, M.; Yaeger, R. Impact of somatic mutations on patterns of metastasis in colorectal cancer. J. Gastrointest. Oncol. 2015, 6, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Elia, I.; Haigis, M.C. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Pratx, G. Imaging metabolic heterogeneity in cancer. Mol. Cancer 2016, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Salomon, A.R.; Voehringer, D.W.; Herzenberg, L.A.; Khosla, C. Understanding and exploiting the mechanistic basis for selectivity of polyketide inhibitors of F0F1-ATPase. Proc. Natl. Acad. Sci. USA 2000, 97, 14766–14771. [Google Scholar] [CrossRef] [PubMed]
- Cottet-Rousselle, C.; Ronot, X.; Leverve, X.; Mayol, J. Cytometric assessment of mitochondria using fluorescent probes. Cytom. Part A 2011, 79A, 405–425. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Mijanovic, O.; Petushkova, A.I.; Brankovic, A.; Turk, B.; Solovieva, A.B.; Nikitkina, A.I.; Bolevich, S.; Timashev, P.S.; Parodi, A.; Zamyatnin, A.A. Cathepsin D—Managing the Delicate Balance. Pharmaceutics 2021, 13, 837. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Steen, H.; Öllinger, K.; Roberg, K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003, 10, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Bidère, N.; Lorenzo, H.K.; Carmona, S.; Laforge, M.; Harper, F.; Dumont, C.; Senik, A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J. Biol. Chem. 2003, 278, 31401–31411. [Google Scholar] [CrossRef] [PubMed]
- Minarowska, A.; Minarowski, Ł.; Karwowska, A.; Gacko, M. Regulatory role of cathepsin D in apoptosis. Folia Histochem. Cytobiol. 2007, 45, 159–163. [Google Scholar]
- Sagulenko, V.; Muth, D.; Sagulenko, E.; Paffhausen, T.; Schwab, M.; Westermann, F. Cathepsin D protects human neuroblastoma cells from doxorubicin-induced cell death. Carcinogenesis 2008, 29, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.-Q.; Han, X.-L.; Kang, X.-L.; Wang, D.; Chen, C.-H.; Wang, J.-X.; Zhao, X.-F. Autophagy triggers CTSD (cathepsin D) maturation and localization inside cells to promote apoptosis. Autophagy 2021, 17, 1170–1192. [Google Scholar] [CrossRef] [PubMed]
- Masson, O.; Bach, A.-S.; Derocq, D.; Prébois, C.; Laurent-Matha, V.; Pattingre, S.; Liaudet-Coopman, E. Pathophysiological functions of cathepsin D: Targeting its catalytic activity versus its protein binding activity? Biochimie 2010, 92, 1635–1643. [Google Scholar] [CrossRef]
- Gyrd-Hansen, M.; Nylandsted, J.; Jäättelä, M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 2004, 3, 1484–1485. [Google Scholar] [CrossRef] [PubMed]
- Leto, G.; Tumminello, F.M.; Crescimanno, M.; Flandina, C.; Gebbia, N. Cathepsin D expression levels in nongynecological solid tumors: Clinical and therapeutic implications. Clin. Exp. Metastasis 2004, 21, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D-Many functions of one aspartic protease. Crit. Rev. Oncol./Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Palermo, C.; Joyce, J.A. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol. Sci. 2008, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Cheriyamundath, S.; Gavert, N.; Brabletz, T.; Haase, G.; Ben-Ze’ev, A. Increased expression of cathepsin D is required for L1-mediated colon cancer progression. Oncotarget 2019, 10, 5217–5228. [Google Scholar] [CrossRef] [PubMed]
- Pereira, H.; Oliveira, C.S.F.; Castro, L.; Preto, A.; Chaves, S.R.; Côrte-Real, M. Yeast as a tool to explore cathepsin D function. Microb. Cell 2015, 2, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Gaisne, M.; Bécam, A.M.; Verdière, J.; Herbert, C.J. A ‘natural’ mutation in Saccharomyces cerevisiae strains derived from S288c affects the complex regulatory gene HAP1 (CYP1). Curr. Genet. 1999, 36, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281–37299. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Thierbach, R.; Voigt, A.; Drewes, G.; Mietzner, B.; Steinberg, P.; Pfeiffer, A.F.H.; Ristow, M. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J. Biol. Chem. 2006, 281, 977–981. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.A. Is Reliance on Mitochondrial Respiration a ‘Chink in the Armor’ of Therapy-Resistant Cancer? Cancer Cell 2014, 26, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.A.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-Fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, S.; Santos-Pereira, C.; Oliveira, C.S.F.; Preto, A.; Chaves, S.R.; Côrte-Real, M. Enhancement of Acetate-Induced Apoptosis of Colorectal Cancer Cells by Cathepsin D Inhibition Depends on Oligomycin A-Sensitive Respiration. Biomolecules 2024, 14, 473. https://doi.org/10.3390/biom14040473
Alves S, Santos-Pereira C, Oliveira CSF, Preto A, Chaves SR, Côrte-Real M. Enhancement of Acetate-Induced Apoptosis of Colorectal Cancer Cells by Cathepsin D Inhibition Depends on Oligomycin A-Sensitive Respiration. Biomolecules. 2024; 14(4):473. https://doi.org/10.3390/biom14040473
Chicago/Turabian StyleAlves, Sara, Cátia Santos-Pereira, Cláudia S. F. Oliveira, Ana Preto, Susana R. Chaves, and Manuela Côrte-Real. 2024. "Enhancement of Acetate-Induced Apoptosis of Colorectal Cancer Cells by Cathepsin D Inhibition Depends on Oligomycin A-Sensitive Respiration" Biomolecules 14, no. 4: 473. https://doi.org/10.3390/biom14040473