
Joint Hypermobility Syndrome and Membrane Proteins: A Comprehensive Review

 , ,
, ,  , and
, and
Abstract
:1. Introduction to Joint Hypermobility Syndrome (JHS) and Its Clinical Manifestations
2. Etiologies of Musculoskeletal Pain and Hypermobility
3. Diagnostic Criteria and Challenges in Diagnosing JHS
4. Proteome Profiling of Membrane Proteins as a Diagnostic Tool for JHS
5. Cellular and Molecular Mechanisms Involved in the Pathogenesis of JHS, with a Focus on Membrane Proteins
5.1. Collagen
5.2. Tenascin XB
5.3. Vitronectin
5.4. Proteoglycan
5.5. Glycosaminoglycan
5.6. Polypetides
6. The Relationship between JHS and Ehlers–Danlos Syndromes/Hypermobility Spectrum Disorders
7. Proteomic Analysis for the Identification of Serum Diagnostic Markers for JHS
8. Mechanisms and Diagnosis of Folate-Dependent Hypermobility Syndrome
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grahame, R. Joint Hypermobility and Genetic Collagen Disorders: Are They Related? Arch. Dis. Child. 1999, 80, 188–191. [Google Scholar] [CrossRef]
- Malfait, F.; Hakim, A.J.; De Paepe, A.; Grahame, R. The Genetic Basis of the Joint Hypermobility Syndromes. Rheumatology 2006, 45, 502–507. [Google Scholar] [CrossRef]
- Castori, M.; Morlino, S.; Celletti, C.; Ghibellini, G.; Bruschini, M.; Grammatico, P.; Blundo, C.; Camerota, F. Re-writing the Natural History of Pain and Related Symptoms in the Joint Hypermobility Syndrome/Ehlers–Danlos Syndrome, Hypermobility Type. Am. J. Med. Genet. Part A Semin. Med. Genet. 2013, 161, 2989–3004. [Google Scholar] [CrossRef]
- Tinkle, B.T.; Levy, H.P. Symptomatic Joint Hypermobility. Med. Clin. N. Am. 2019, 103, 1021–1033. [Google Scholar] [CrossRef]
- Remvig, L.; Jensen, D.V.; Ward, R.C. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: Review of the literature. J. Rheumatol. 2007, 34, 804–809. [Google Scholar]
- Castori, M.; Hakim, A. Contemporary approach to joint hypermobility and related disorders. Curr. Opin. Pediatr. 2017, 29, 640–649. [Google Scholar] [CrossRef]
- Simpson, M.R. Benign Joint Hypermobility Syndrome: Evaluation, Diagnosis, and Management. J. Am. Osteopath. Assoc. 2006, 106, 531–536. [Google Scholar]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers–Danlos syndrome (aka Ehlers–Danlos syndrome Type III and Ehlers–Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef]
- Castori, M.; Tinkle, B. A framework for the classification of joint hypermobility and related conditions. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 148–157. [Google Scholar] [CrossRef]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 6–22. [Google Scholar] [CrossRef]
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The ehlers–danlos syndromes. Nat. Rev. Dis. Primers 2020, 6, 64. [Google Scholar] [CrossRef]
- Sacheti, A.; Szemere, J.; Bernstein, B.; Tafas, T.; Schechter, N.; Tsipouras, P. Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J. Pain Symptom Manag. 1997, 14, 88–93. [Google Scholar] [CrossRef]
- Hakim, A.; Grahame, R. Joint hypermobility. Best Pract. Res. Clin. Rheumatol. 2003, 17, 989–1004. [Google Scholar] [CrossRef]
- Castori, M.; Morlino, S.; Celletti, C.; Celli, M.; Morrone, A.; Colombi, M.; Grammatico, P. Management of pain and fatigue in the joint hypermobility syndrome (aka Ehlers–Danlos syndrome, hypermobility type): Principles and proposal for a multidisciplinary approach. Am. J. Med. Genet. Part A Semin. Med. Genet. 2012, 158, 2055–2070. [Google Scholar] [CrossRef]
- Volberding, P.A.; Spicer, C.M.; Cartaxo, T.; Wedge, R.A. Selected Heritable Disorders of Connective Tissue and Disability; National Academies Press: Washington, DC, USA, 2022; ISBN 9780309275538. [Google Scholar]
- Gensemer, C.; Burks, R.; Kautz, S.; Judge, D.P.; Lavallee, M.; Norris, R.A. Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Dev. Dyn. 2021, 250, 318–344. [Google Scholar] [CrossRef]
- Punzi, L.; Pozzuoli, A.; Pianon, M.; Bertazzolo, N.; Oliviero, F.; Scapinelli, R. Pro-inflammatory interleukins in the synovial fluid of rheumatoid arthritis associated with joint hypermobility. Rheumatology 2001, 40, 202–204. [Google Scholar] [CrossRef]
- Rodgers, K.; Gui, J.; Dinulos, M.; Chou, R. Ehlers-Danlos syndrome hypermobility type is associated with rheumatic diseases. Sci. Rep. 2017, 7, 39636. [Google Scholar] [CrossRef]
- Gavrilova, N.; Soprun, L.; Lukashenko, M.; Ryabkova, V.; Fedotkina, T.V.; Churilov, L.P.; Shoenfeld, Y. New Clinical Phenotype of the Post-Covid Syndrome: Fibromyalgia and Joint Hypermobility Condition. Pathophysiology 2022, 29, 24–29. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 International Classification of the Ehlers–Danlos Syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef]
- Malfait, F.; Colman, M.; Vroman, R.; De Wandele, I.; Rombaut, L.; Miller, R.E.; Syx, D. Pain in the Ehlers–Danlos syndromes: Mechanisms, models, and challenges. Am. J. Med. Genet. Part C Semin. Med. Genet. 2021, 187, 429–445. [Google Scholar] [CrossRef]
- Chiarelli, N.; Zoppy, N. Matrix Metalloproteinases Inhibition by Doxycycline Rescues Extracellular Matrix Organization and Partly Reverts Myofibroblast Differentiation in Hypermobile Ehlers-Danlos Syndrome Dermal Fibroblasts: A Potential Therapeutic Target? Cells 2021, 10, 3236. [Google Scholar] [CrossRef]
- Ritelli, M.; Chiarelli, N.; Cinquina, V.; Vezzoli, M.; Venturini, M.; Colombi, M. Looking back and beyond the 2017 diagnostic criteria for hypermobile Ehlers-Danlos syndrome: A retrospective cross-sectional study from an Italian reference center. Am. J. Med. Genet. Part A Semin. Med. Genet. 2024, 194, 174–194. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N. RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms. Cells 2022, 11, 4040. [Google Scholar] [CrossRef] [PubMed]
- Cattalini, M.; Khubchandani, R.; Cimaz, R. When flexibility is not necessarily a virtue: A review of hypermobility syndromes and chronic or recurrent musculoskeletal pain in children. Pediatr. Rheumatol. 2015, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.B. Hypermobility spectrum disorders: A review. Rheumatol. Immunol. Res. 2023, 4, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Lenert, P. Joint hypermobility syndrome: Recognizing a commonly overlooked cause of chronic pain. Am. J. Med. 2017, 130, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Tofts, L.J.; Elliott, E.J.; Munns, C.; Pacey, V.; Sillence, D.O. The differential diagnosis of children with joint hypermobility: A review of the literature. Pediatr. Rheumatol. 2009, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Malek, S.; Reinhold, E.J.; Pearce, G.S. The Beighton Score as a measure of generalised joint hypermobility. Rheumatol. Int. 2021, 41, 1707–1716. [Google Scholar] [CrossRef]
- Engelbert, R.H.; Rombaut, L. Clinimetrics: Assessment of generalised joint hypermobility: The Beighton score. J. Physiother. 2022, 68, 208. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Dordoni, C.; Ritelli, M.; Venturini, M.; Venturini, M.; Castori, M.; Colombi, M. Transcriptome-Wide Expression Profiling in Skin Fibroblasts of Patients with Joint Hypermobility Syndrome/Ehlers-Danlos Syndrome Hypermobility Type. PLoS ONE 2016, 11, e0161347. [Google Scholar] [CrossRef]
- Chiarelli, N.; Ritelli, M.; Zoppi, N.; Colombi, M. Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers-Danlos Syndromes. Genes 2019, 10, 609. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, N.; Zoppi, N.; Ritelli, M.; Venturini, M.; Capitanio, D.; Gelfi, C.; Colombi, M. Biological insights in the pathogenesis of hypermobile Ehlers-Danlos syndrome from proteome profiling of patients’ dermal myofibroblasts. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166051. [Google Scholar] [CrossRef] [PubMed]
- Malek, S.; Köster, D.V. The Role of Cell Adhesion and Cytoskeleton Dynamics in the Pathogenesis of the Ehlers-Danlos Syndromes and Hypermobility Spectrum Disorders. Front. Cell Dev. Biol. 2021, 9, 649082. [Google Scholar] [CrossRef] [PubMed]
- de Jong, E.; Kocer, A. Current Methods for Identifying Plasma Membrane Proteins as Cancer Biomarkers. Membranes 2023, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qin, H.; Ye, M. An overview on enrichment methods for cell surface proteome profiling. J. Sep. Sci. 2020, 43, 292–312. [Google Scholar] [CrossRef]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB Is Associated with Hypermobility Type of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Satoh, K.; Maniwa, T.; Matsumoto, K.I. Proteomic analysis for the identification of serum diagnostic markers for joint hypermobility syndrome. Int. J. Mol. Med. 2016, 37, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Kosho, T.; Matsumoto, N. Ehlers–Danlos syndrome associated with glycosaminoglycan abnormalities. In Progress in Heritable Soft Connective Tissue Diseases; Springer: Dordrecht, The Netherlands, 2014; pp. 145–159. [Google Scholar]
- Castori, M.; Colombi, M. Generalized Joint Hypermobility, Joint Hypermobility Syndrome and Ehlers-Danlos Syndrome, Hypermobility Type. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, R. The Human Collagen Mutation Database 1998. Nucleic Acids Res. 1998, 26, 253–255. [Google Scholar] [CrossRef]
- Dalgleish, R. The Human Type I Collagen Mutation Database. Nucleic Acids Res. 1997, 25, 181–187. [Google Scholar] [CrossRef]
- Byers, P.H.; Pyeritz, R.E.; Uitto, J. Research Perspectives in Heritable Disorders of Connective Tissue. Matrix 1992, 12, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Franzke, C.-W.; Bruckner, P.; Bruckner-Tuderman, L. Collagenous Transmembrane Proteins: Recent Insights into Biology and Pathology. J. Biol. Chem. 2005, 280, 4005–4008. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Steplewski, A.; Chung, K.Y.; Uitto, J.; Fertala, A. Collagen Fibril Formation. J. Biol. Chem. 2008, 283, 25879–25886. [Google Scholar] [CrossRef] [PubMed]
- Heino, J. The Collagen Family Members as Cell Adhesion Proteins. BioEssays 2007, 29, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin Ligands at a Glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B.; Hohenester, E. Mammalian Collagen Receptors. Matrix Biol. 2007, 26, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, M.; Vandersteen, A.; Yiş, U.; Serdaroglu, G.; Ataman, E.; Chopra, M.; Garcia, S.; Jones, K.; Kariminejad, A.; Kraenzlin, M.; et al. Phenotypic Variability of the Kyphoscoliotic Type of Ehlers-Danlos Syndrome (EDS VIA): Clinical, Molecular and Biochemical Delineation. Orphanet J. Rare Dis. 2011, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A Recessive Form of the Ehlers–Danlos Syndrome Caused by Tenascin-X Deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Zweers, M.C.; van Vlijmen-Willems, I.M.; van Kuppevelt, T.H.; Mecham, R.P.; Steijlen, P.M.; Bristow, J.; Schalkwijk, J. Deficiency of Tenascin-X Causes Abnormalities in Dermal Elastic Fiber Morphology. J. Investig. Dermatol. 2004, 122, 885–891. [Google Scholar] [CrossRef]
- Mao, J.R.; Taylor, G.; Dean, W.B.; Wagner, D.R.; Afzal, V.; Lotz, J.C.; Rubin, E.M.; Bristow, J. Tenascin-X Deficiency Mimics Ehlers-Danlos Syndrome in Mice through Alteration of Collagen Deposition. Nat. Genet. 2002, 30, 421–425. [Google Scholar] [CrossRef]
- Jones, F.S.; Jones, P.L. The Tenascin Family of ECM Glycoproteins: Structure, Function, and Regulation during Embryonic Development and Tissue Remodeling. Dev. Dyn. 2000, 218, 235–259. [Google Scholar] [CrossRef]
- Speiser, P.W.; White, P.C. Congenital Adrenal Hyperplasia. N. Engl. J. Med. 2003, 349, 776–788. [Google Scholar] [CrossRef]
- Elefteriou, F.; Exposito, J.-Y.; Garrone, R.; Lethias, C. Binding of Tenascin-X to Decorin. FEBS Lett. 2001, 495, 44–47. [Google Scholar] [CrossRef]
- Bristow, J.; Tee, M.; Gitelman, S.; Mellon, S.; Miller, W. Tenascin-X: A Novel Extracellular Matrix Protein Encoded by the Human XB Gene Overlapping P450c21B. J. Cell Biol. 1993, 122, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, I.; Seger, D.; Shaltiel, S. Vitronectin. Int. J. Biochem. Cell Biol. 1999, 31, 539–544. [Google Scholar] [CrossRef]
- Preissner, K.T. Structure and Biological Role of Vitronectin. Annu. Rev. Cell Biol. 1991, 7, 275–310. [Google Scholar] [CrossRef] [PubMed]
- Leavesley, D.I.; Kashyap, A.S.; Croll, T.; Sivaramakrishnan, M.; Shokoohmand, A.; Hollier, B.G.; Upton, Z. Vitronectin—Master Controller or Micromanager? IUBMB Life 2013, 65, 807–818. [Google Scholar] [CrossRef]
- Tollefsen, D.M.; Weigel, C.J.; Kabeer, M.H. The Presence of Methionine or Threonine at Position 381 in Vitronectin Is Correlated with Proteolytic Cleavage at Arginine 379. J. Biol. Chem. 1990, 265, 9778–9781. [Google Scholar] [CrossRef]
- Chillakuri, C.R.; Jones, C.; Mardon, H.J. Heparin Binding Domain in Vitronectin Is Required for Oligomerization and Thus Enhances Integrin Mediated Cell Adhesion and Spreading. FEBS Lett. 2010, 584, 3287–3291. [Google Scholar] [CrossRef]
- Vasilyev, D.V.; Barish, M.E. Regulation of an Inactivating Potassium Current (IA) by the Extracellular Matrix Protein Vitronectin in Embryonic Mouse Hippocampal Neurones. J. Physiol. 2003, 547, 859–871. [Google Scholar] [CrossRef]
- Milis, L.; Morris, C.A.; Sheehan, M.C.; Charlesworth, J.A.; Pussell, B.A. Vitronectin-Mediated Inhibition of Complement: Evidence for Different Binding Sites for C5b-7 and C9. Clin. Exp. Immunol. 2008, 92, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Shimo-Oka, T.; Morishita, N.; Ii, I.; Hayashi, M. Identification of the Collagen-Binding Domain of Vitronectin Using Monoclonal Antibodies. Cell Struct. Funct. 1988, 13, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R.; Preissner, K.T.; Müller-Eberhard, H.J. Inhibition of C9 Polymerization within the SC5b-9 Complex of Complement by S-Protein. Acta Pathol. Microbiol. Immunol. Scand. Suppl. 1984, 284, 89–96. [Google Scholar]
- Podack, E.R.; Kolb, W.P.; Müller-Eberhard, H.J. The SC5b-7 Complex: Formation, Isolation, Properties, and Subunit Composition. J. Immunol. 1977, 119, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; De Paepe, A. The Ehlers-Danlos Syndrome. In Progress in Heritable Soft Connective Tissue Diseases; Springer: Dordrecht, The Netherland, 2014; pp. 129–143. [Google Scholar]
- Bülow, H.E.; Hobert, O. The Molecular Diversity of Glycosaminoglycans Shapes Animal Development. Annu. Rev. Cell Dev. Biol. 2006, 22, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.E. Proteoglycan-Fibrillar Collagen Interactions. Biochem. J. 1988, 252, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.E. Elasticity in Extracellular Matrix ‘Shape Modules’ of Tendon, Cartilage, Etc. A Sliding Proteoglycan-filament Model. J. Physiol. 2003, 553, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H.; Rosthøj, S.; Quentin, E.; Hollmann, J.; Glössl, J.; Okada, S.; Tønnesen, T. Glycosaminoglycan-Free Small Proteoglycan Core Protein Is Secreted by Fibroblasts from a Patient with a Syndrome Resembling Progeroid. Am. J. Hum. Genet. 1987, 41, 436–453. [Google Scholar] [PubMed]
- Ermert, D.; Blom, A.M. C4b-Binding Protein: The Good, the Bad and the Deadly. Novel Functions of an Old Friend. Immunol. Lett. 2016, 169, 82–92. [Google Scholar] [CrossRef]
- Olivar, R.; Luque, A.; Naranjo-Gómez, M.; Quer, J.; García de Frutos, P.; Borràs, F.E.; Rodríguez de Córdoba, S.; Blom, A.M.; Aran, J.M. The A7β0 Isoform of the Complement Regulator C4b-Binding Protein Induces a Semimature, Anti-Inflammatory State in Dendritic Cells. J. Immunol. 2013, 190, 2857–2872. [Google Scholar] [CrossRef]
- Trouw, L.A.; Bengtsson, A.A.; Gelderman, K.A.; Dahlbäck, B.; Sturfelt, G.; Blom, A.M. C4b-Binding Protein and Factor H Compensate for the Loss of Membrane-Bound Complement Inhibitors to Protect Apoptotic Cells against Excessive Complement Attack. J. Biol. Chem. 2007, 282, 28540–28548. [Google Scholar] [CrossRef]
- Trouw, L.A.; Nilsson, S.C.; Gonçalves, I.; Landberg, G.; Blom, A.M. C4b-Binding Protein Binds to Necrotic Cells and DNA, Limiting DNA Release and Inhibiting Complement Activation. J. Exp. Med. 2005, 201, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Wedge, R.A.; Cartaxo, T.; Spicer, C.M.; Volberding, P.A.; National Academies of Sciences, Engineering, and Medicine. Ehlers-Danlos Syndromes and Hypermobility Spectrum Disorders. In Selected Heritable Disorders of Connective Tissue and Disability; National Academies Press: Washington, DC, USA, 2022. [Google Scholar]
- Yew, K.S.; Kamps-Schmitt, K.A.; Borge, R. Hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Am. Fam. Physician 2021, 103, 481–492. [Google Scholar] [PubMed]
- Thompson, A.; Danesh, J. Associations between apolipoprotein B, apolipoprotein AI, the apolipoprotein B/AI ratio and coronary heart disease: A literature-based meta-analysis of prospective studies. J. Intern. Med. 2006, 259, 481–492. [Google Scholar] [CrossRef]
- Mercado, M.; Salgado-Goytia, L. Increase levels of apo-A1 and apo B are asso¬ciated in knee osteoarthritis: Lack of association with VEGF-460 T/C and +405 C/G polymorphisms. Rheumatol. Int. 2008, 29, 63–68. [Google Scholar]
- Monaco, H.L. The transthyretin-retinol-binding protein complex. Biochim. Biophys. Acta (BBA) 2000, 1482, 65–72. [Google Scholar] [CrossRef]
- Wilson, G.N. Exome analysis of connective tissue dysplasia: Death and rebirth of clinical genetics? Am. J. Med. Genet. Part A Semin. Med. Genet. 2014, 164A, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Merke, D.P.; Chen, W. Tenascin-X Haploinsufficiency Associated with Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J. Clin. Endoocrinol. Metab. 2013, 98, E379–E387. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Watanabe, A. A method for quantification of serum tenascin-X by nano-LC/MS/MS. Clin. Chim. Acta 2016, 459, 94–100. [Google Scholar] [CrossRef]
- Yamada, K.; Watanabe, A. Measurement of Serum Tenascin-X in Joint Hypermobility Syndrome Patients. Biol. Farm. Bull. 2019, 42, 1596–1599. [Google Scholar] [CrossRef]
- Schneider, M.; Hansen, J.L. S100A4: A common mediator of epithelial-mesenchymal transition, fibrosis and regeneration in diseases? J. Mol. Med. 2008, 86, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y. Extracellular S100A4 as a key player in fibrotic diseases. J. Cell. Mol. Med. 2020, 24, 5973–5983. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Li, F. S100A4 amplifies TGF- β-induced epithelial–mesenchymal transition in a pleural mesothelial cell line. J. Investig. Med. 2018, 66, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Qu, J. Role of metastasis-induced protein S100A4 in human non-tumor pathophysiologies. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Ambartsumian, N.; Klingelhöfer, J. The multifaceted S100A4 protein in cancer and inflammation. Methods Mol. Biol. 2019, 1929, 339–365. [Google Scholar] [PubMed]
- Garrett, S.C.; Varney, K.M. S100A4, a mediator of metastasis. J. Biol. Chem. 2006, 281, 677–680. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R. Functions of S100 proteins. Curr. Mol. Med. 2012, 13, 24–57. [Google Scholar] [CrossRef]
- Saleem, M.; Kweon, M.H. S100A4 accelerates tumorigenesis and invasion of human prostate cancer through the transcriptional regulation of matrix metalloproteinase 9. Proc. Natl. Acad. Sci. USA 2006, 103, 14825–14830. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, L.E.; RemáKová, M. The metastasisassociated protein S100A4 promotes the inflammatory response of mononuclear cells via the TLR4 signaling pathway in rheumatoid arthritis. Rheumatology 2014, 53, 1520–1526. [Google Scholar] [CrossRef]
- Chaabane, C.; Heizmann, C.W. Extracellular S100A4 induces smooth muscle cell phenotypic transition mediated by RAGE. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2144–2157. [Google Scholar]
- Schmidt-Hansen, B.; Ornås, D. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene 2004, 23, 5487–5495. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Chiarelli, N. Dermal fibroblast-tomyofibroblast transition sustained by αvß3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Caliogna, L.; Guerrieri, V. Biomarkers for Ehlers-Danlos Syndromes: There Is a Role? Int. J. Mol. Sci. 2021, 22, 10149. [Google Scholar] [CrossRef] [PubMed]
- Glatt, S.J.; Everall, I.P. Comparative Gene Expression Analysis of Blood and Brain Provides Concurrent Validation of SELENBP1 Up-Regulation in Schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 15533–15538. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, M.; Celletti, C. Unexpected Association between Joint Hypermobility Syndrome/Ehlers-Danlos Syndrome Hypermobility Type and Obsessive-Compulsive Personality Disorder. Rheumatol. Int. 2014, 34, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Hershenfeld, S.A.; Wasim, S. Psychiatric Disorders in Ehlers-Danlos Syndrome Are Frequent, Diverse and Strongly Associated with Pain. Rheumatol. Int. 2016, 36, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Courseault, J.; Kingry, C.; Morrison, V.; Edstrom, C.; Morrell, K.; Jaubert, L.; Elia, V.; Bix, G. Folate-Dependent Hypermobility Syndrome: A Proposed Mechanism and Diagnosis. Heliyon 2023, 9, e15387. [Google Scholar] [CrossRef] [PubMed]
- Guéant-Rodriguez, R.M.; Guéant, J.L.; Debard, R.; Thirion, S.; Lu, X.H.; Bronowicki, J.P.; Namour, F.; Chabi, N.W.; Sanni, A.; Anello, G.; et al. Prevalence of Methylenetetrahydrofolate Reductase 677T and 1298C Alleles and Folate Status: A Comparative Study in Mexican, West African, and European Populations. Am. J. Clin. Nutr. 2006, 83, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Tsang, B.L.; Devine, O.J.; Cordero, A.M.; Marchetta, C.M.; Mulinare, J.; Mersereau, P.; Guo, J.; Qi, Y.P.; Berry, R.J.; Rosenthal, J.; et al. Assessing the Association between the Methylenetetrahydrofolate Reductase (MTHFR) 677>T Polymorphism and Blood Folate Concentrations: A Systematic Review and Meta-Analysis of Trials and Observational Studies. Am. J. Clin. Nutr. 2015, 101, 1286–1294. [Google Scholar] [CrossRef]
- Ni, J.; Zhang, L.; Zhou, T.; Xu, W.J.; Xue, J.L.; Cao, N.; Wang, X. Association between the MTHFR C677T Polymorphism, Blood Folate and Vitamin B12 Deficiency, and Elevated Serum Total Homocysteine in Healthy Individuals in Yunnan Province, China. J. Chin. Med. Assoc. 2017, 80, 147–153. [Google Scholar] [CrossRef]
- Li, W.X.; Dai, S.X.; Zheng, J.J.; Liu, J.Q.; Huang, J.F. Homocysteine Metabolism Gene Polymorphisms (MTHFR C677T, MTHFR A1298C, MTR A2756G and MTRR A66G) Jointly Elevate the Risk of Folate Deficiency. Nutrients 2015, 7, 6670–6687. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zeng, Y.; Pei, P.; He, X.; Liu, F.; Zhang, T. Abnormal Transcriptome-Wide DNA Demethylation Induced by Folate Deficiency Causes Neural Tube Defects. Front. Genet. 2022, 13, 987210. [Google Scholar] [CrossRef] [PubMed]
- Gubbiotti, M.A.; Vallet, S.D.; Ricard-Blum, S.; Iozzo, R.V. Decorin Interacting Network: A Comprehensive Analysis of Decorin-Binding Partners and Their Versatile Functions. Matrix Biol. 2016, 55, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin Is a Pivotal Effector in the Extracellular Matrix and Tumour Microenvironment. Oncotarget 2018, 9, 5480. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Simons, M. TGFβ Signaling Pathways in Human Health and Disease. Front. Mol. Biosci. 2023, 10, 1113061. [Google Scholar] [CrossRef]
- O’Connor, C.; Wallace-Povirk, A.; Ning, C.; Frühauf, J.; Tong, N.; Gangjee, A.; Matherly, L.H.; Hou, Z. Folate Transporter Dynamics and Therapy with Classic and Tumor-Targeted Antifolates. Sci. Rep. 2021, 11, 6389. [Google Scholar] [CrossRef] [PubMed]
- Bobrowski-Khoury, N.; Ramaekers, V.T.; Sequeira, J.M.; Quadros, E.V. Folate Receptor Alpha Autoantibodies in Autism Spectrum Disorders: Diagnosis, Treatment and Prevention. J. Pers. Med. 2021, 11, 710. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Cerebral Folate Deficiency, Folate Receptor Alpha Autoantibodies and Leucovorin (Folinic Acid) Treatment in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. J. Pers. Med. 2021, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, J. The Evolution of Folate Supplementation—From One Size for All to Personalized, Precision, Poly-Paths. J. Transl. Intern. Med. 2023, 11, 128–137. [Google Scholar] [CrossRef]
- He, J.; Qin, M.; Chen, Y.; Hu, Z.; Xie, F.; Ye, L.; Hui, T. Epigenetic Regulation of Matrix Metalloproteinases in Inflammatory Diseases: A Narrative Review. Cell Biosci. 2020, 10, 86. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Quadros, E.V. Cerebral Folate Deficiency Syndrome: Early Diagnosis, Intervention and Treatment Strategies. Nutrients 2022, 14, 3096. [Google Scholar] [CrossRef] [PubMed]
- Kindgren, E.; Perez, A.Q.; Knez, R. Prevalence of Adhd and Autism Spectrum Disorder in Children with Hypermobility Spectrum Disorders or Hypermobile Ehlers-Danlos Syndrome: A Retrospective Study. Neuropsychiatr. Dis. Treat. 2021, 17, 379–388. [Google Scholar] [CrossRef] [PubMed]
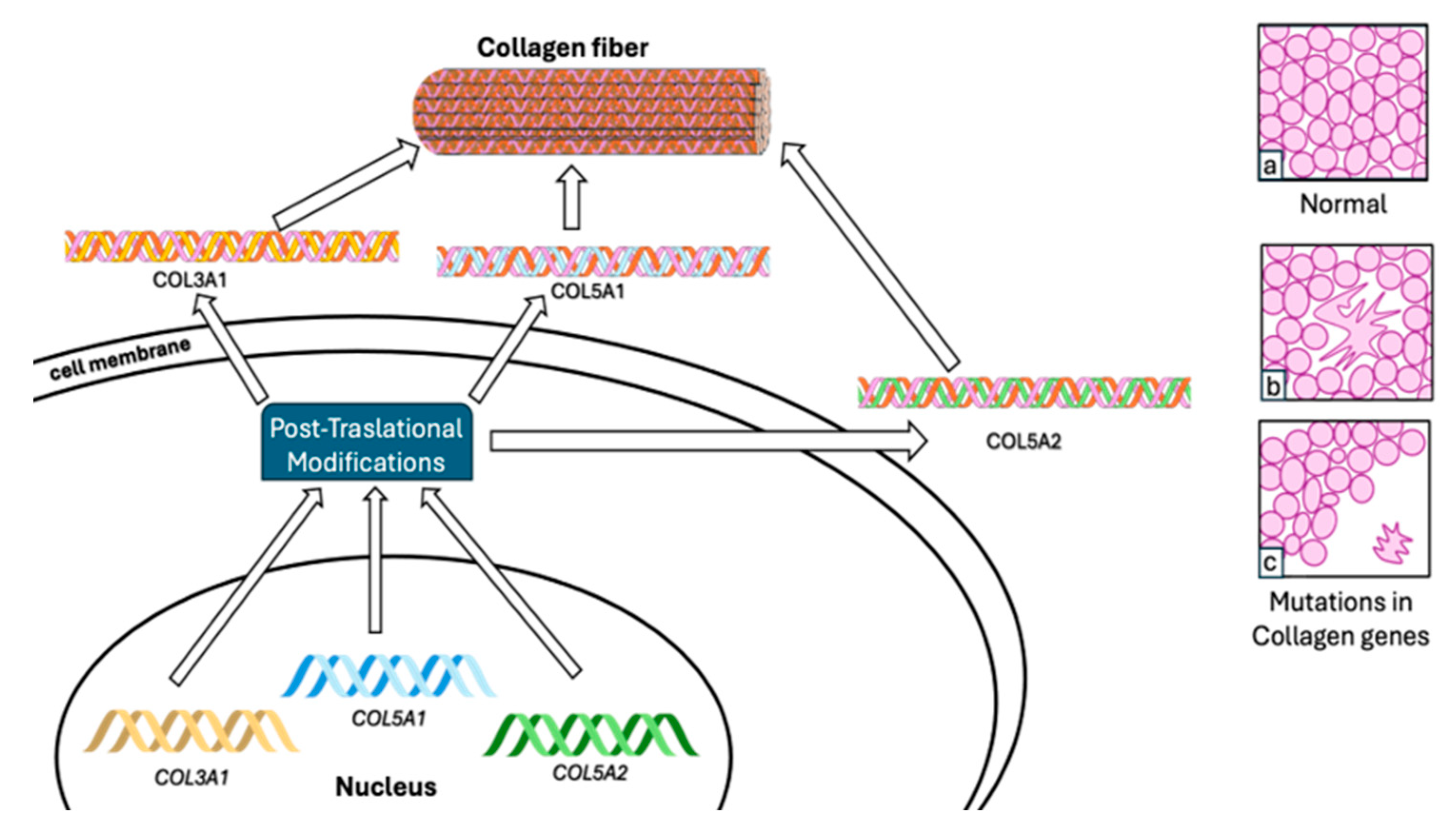


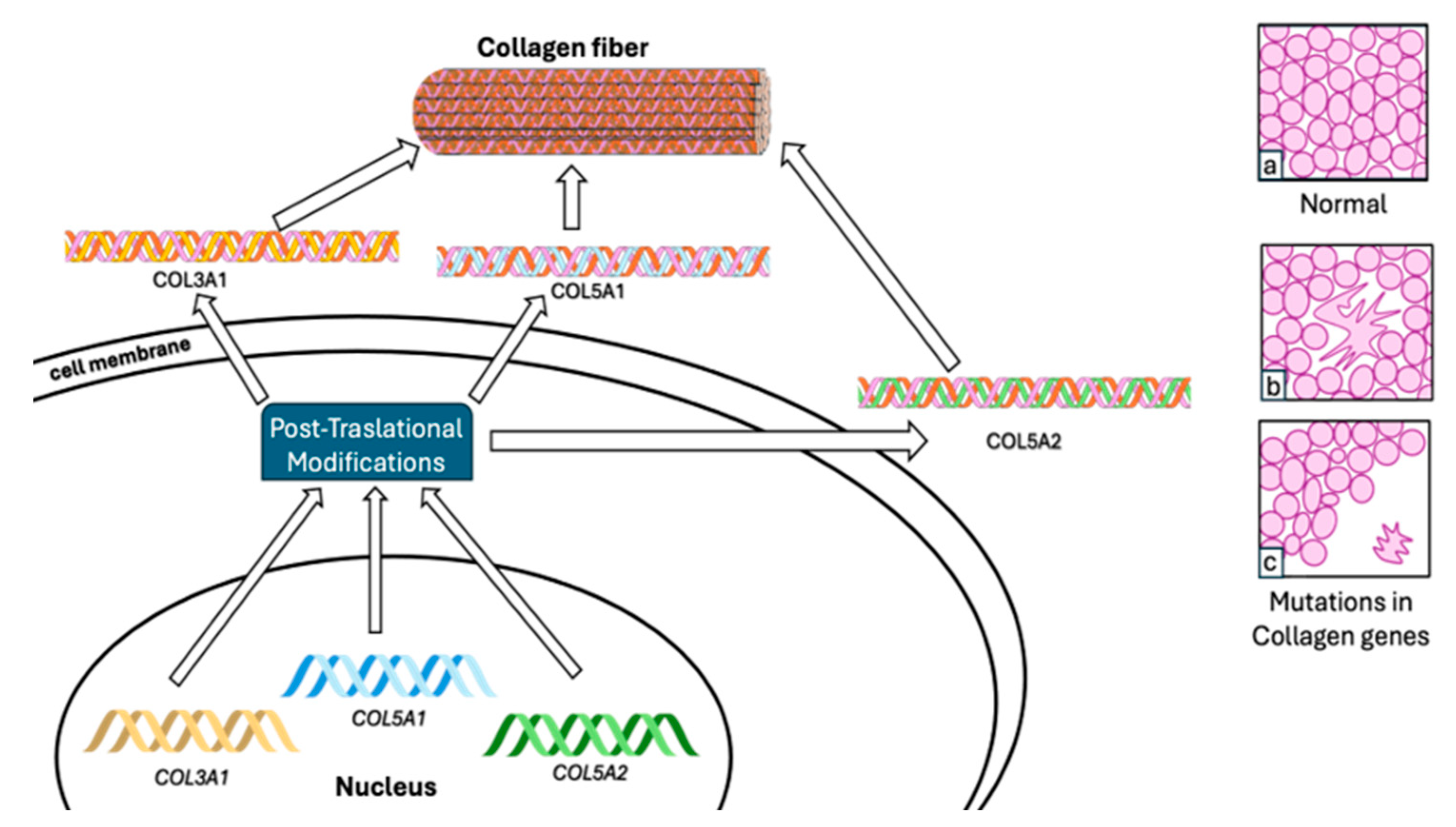{kind=link}
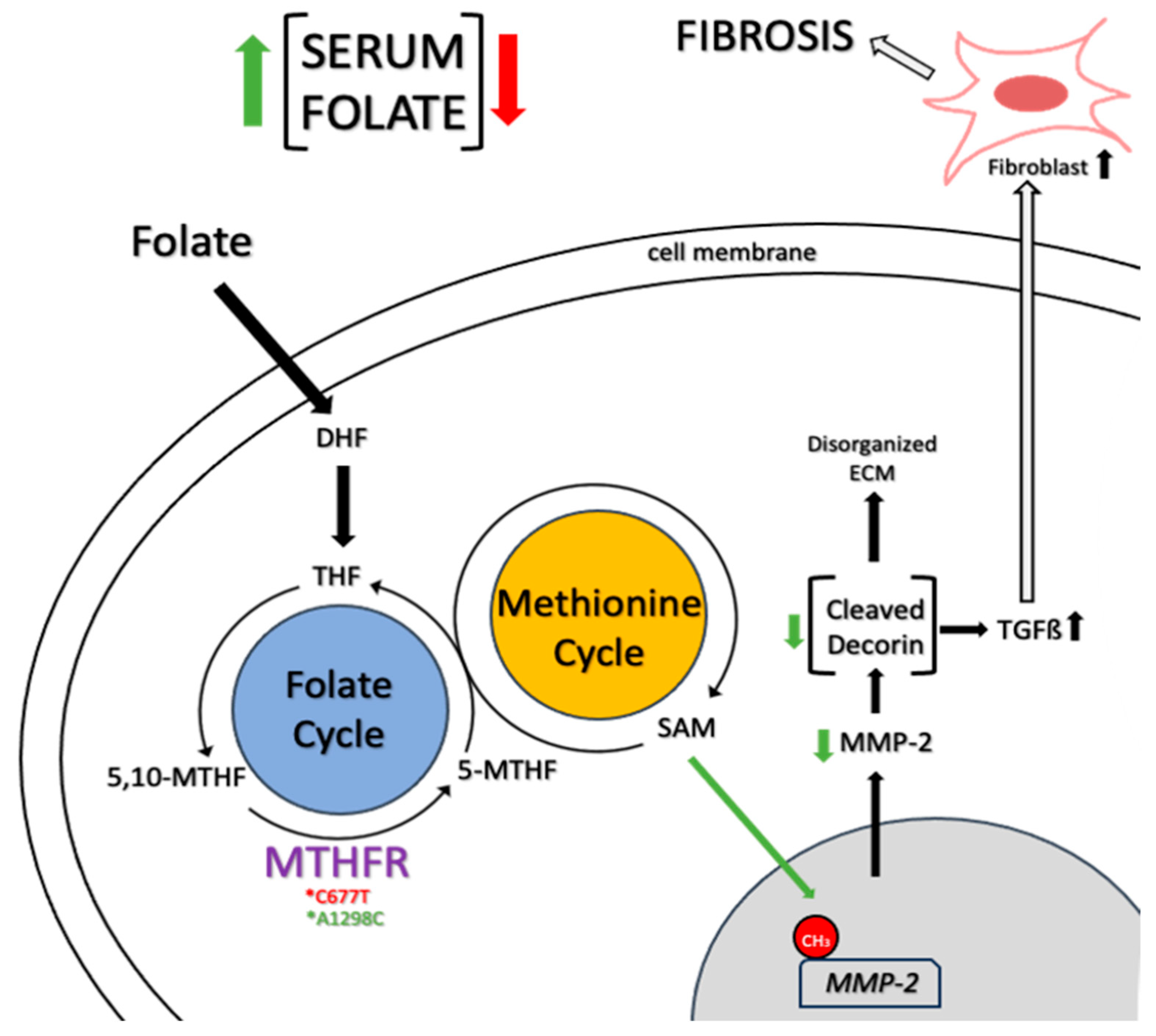{kind=link}
| Biomarker Candidates | Expression Landscape | Sample Type | Reference |
|---|---|---|---|
| Proteins involved with the complement system: C1r, C1R, VTN, C14, and C4BPA. | Increased for JHS | Human serum | [4] |
| Apolipoprotein B-100 (APOB) | Increased for JHS | Human serum | [4,7] |
| Transthyretin (TTR) | Increased for JHS | Human serum | [4,37] |
| Protein tenascin-X | Reduced for cEDS and hEDS | Human serum | [4,37] |
| S100A4 | Increased for hEDS | Dermal myofibroblasts | [39,47] |
| Interleukin 6 | Reduced for hEDS | Fibroblasts | [48,49] |
| Prolactin | Possibly increased for EDS | Fibroblasts | [48,49] |
| Selenium-binding protein-1 (SELENBP1) | Possibly increased for hEDS and schizophrenia | Blood and brain | [49,50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pliego-Arreaga, R.; Cervantes-Montelongo, J.A.; Silva-Martínez, G.A.; Tristán-Flores, F.E.; Pantoja-Hernández, M.A.; Maldonado-Coronado, J.R. Joint Hypermobility Syndrome and Membrane Proteins: A Comprehensive Review. Biomolecules 2024, 14, 472. https://doi.org/10.3390/biom14040472
Pliego-Arreaga R, Cervantes-Montelongo JA, Silva-Martínez GA, Tristán-Flores FE, Pantoja-Hernández MA, Maldonado-Coronado JR. Joint Hypermobility Syndrome and Membrane Proteins: A Comprehensive Review. Biomolecules. 2024; 14(4):472. https://doi.org/10.3390/biom14040472
Chicago/Turabian StylePliego-Arreaga, Raquel, Juan Antonio Cervantes-Montelongo, Guillermo Antonio Silva-Martínez, Fabiola Estefanía Tristán-Flores, Miguel Angel Pantoja-Hernández, and Juan Raúl Maldonado-Coronado. 2024. "Joint Hypermobility Syndrome and Membrane Proteins: A Comprehensive Review" Biomolecules 14, no. 4: 472. https://doi.org/10.3390/biom14040472
APA StylePliego-Arreaga, R., Cervantes-Montelongo, J. A., Silva-Martínez, G. A., Tristán-Flores, F. E., Pantoja-Hernández, M. A., & Maldonado-Coronado, J. R. (2024). Joint Hypermobility Syndrome and Membrane Proteins: A Comprehensive Review. Biomolecules, 14(4), 472. https://doi.org/10.3390/biom14040472