_Kwok.png)
Recent Advances on Mutant p53: Unveiling Novel Oncogenic Roles, Degradation Pathways, and Therapeutic Interventions

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Novel Mechanisms of Mutant p53 Gain of Function (GOF)
2.1. Metabolic Regulations by Mutant p53
2.2. Mutant p53 Sustains the Warburg Effect Thus Promoting Lactate Secretion
2.3. Cancer Secretome Regulation by Mutp53
2.4. Mutp53 Regulation of TME and Tumor-Stroma Communication via Extracellular Vesicles (EVs)
2.5. Other Novel Mechanisms by Which Mutp53 Promotes Cancer Cell Invasion and Metastasis
2.6. Sirtuin/P53 Axis: Interplay and Potential Speculation
3. Mechanisms of Mutp53 Degradation
3.1. Mutant p53 Degradation: The Proteasomal Route
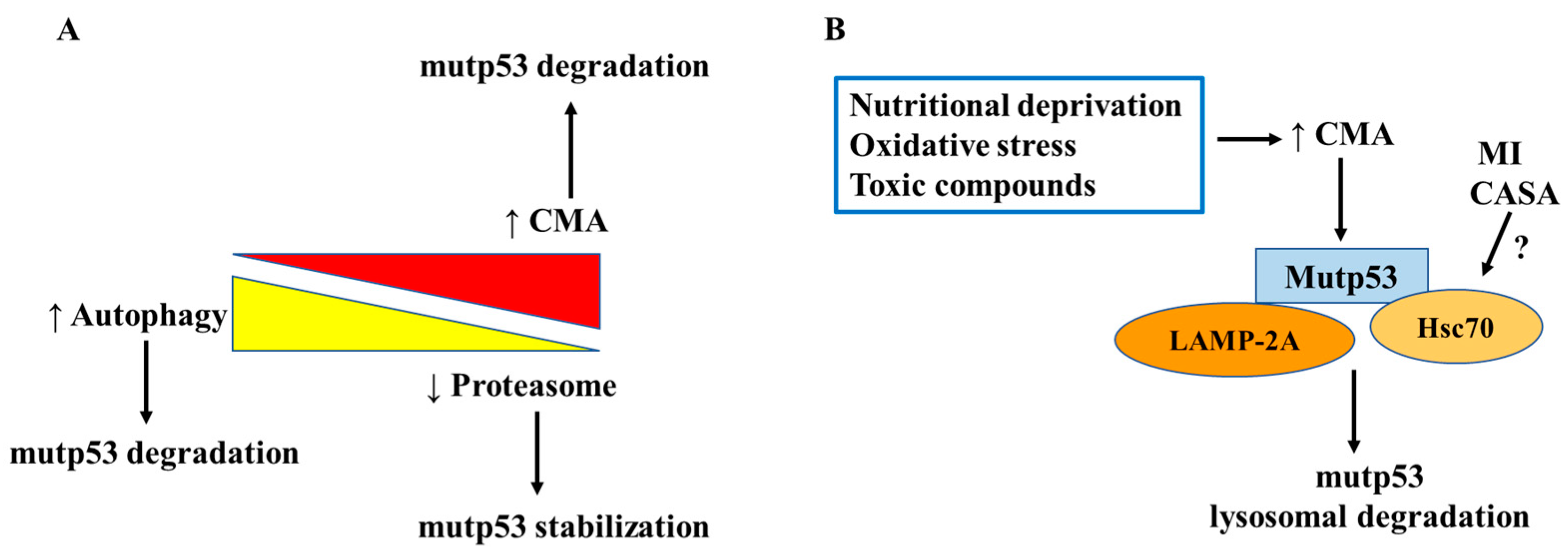
3.2. Mutant p53 Degradation: The Lysosomal Routes
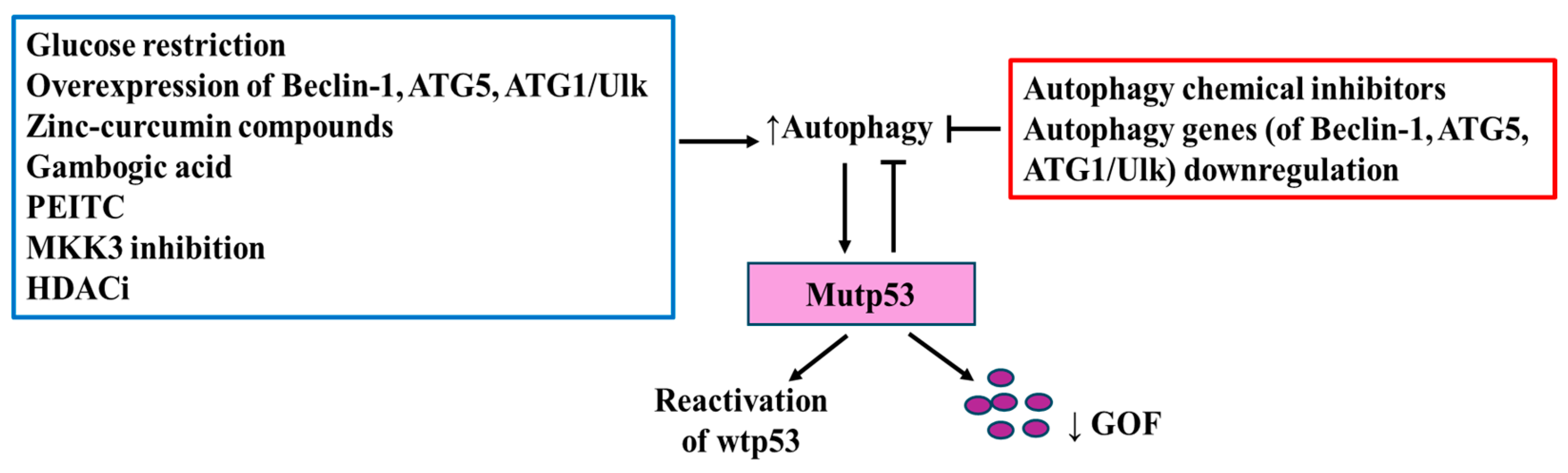
3.3. Autophagy Regulation by Mutant p53 and Its Impact on TME
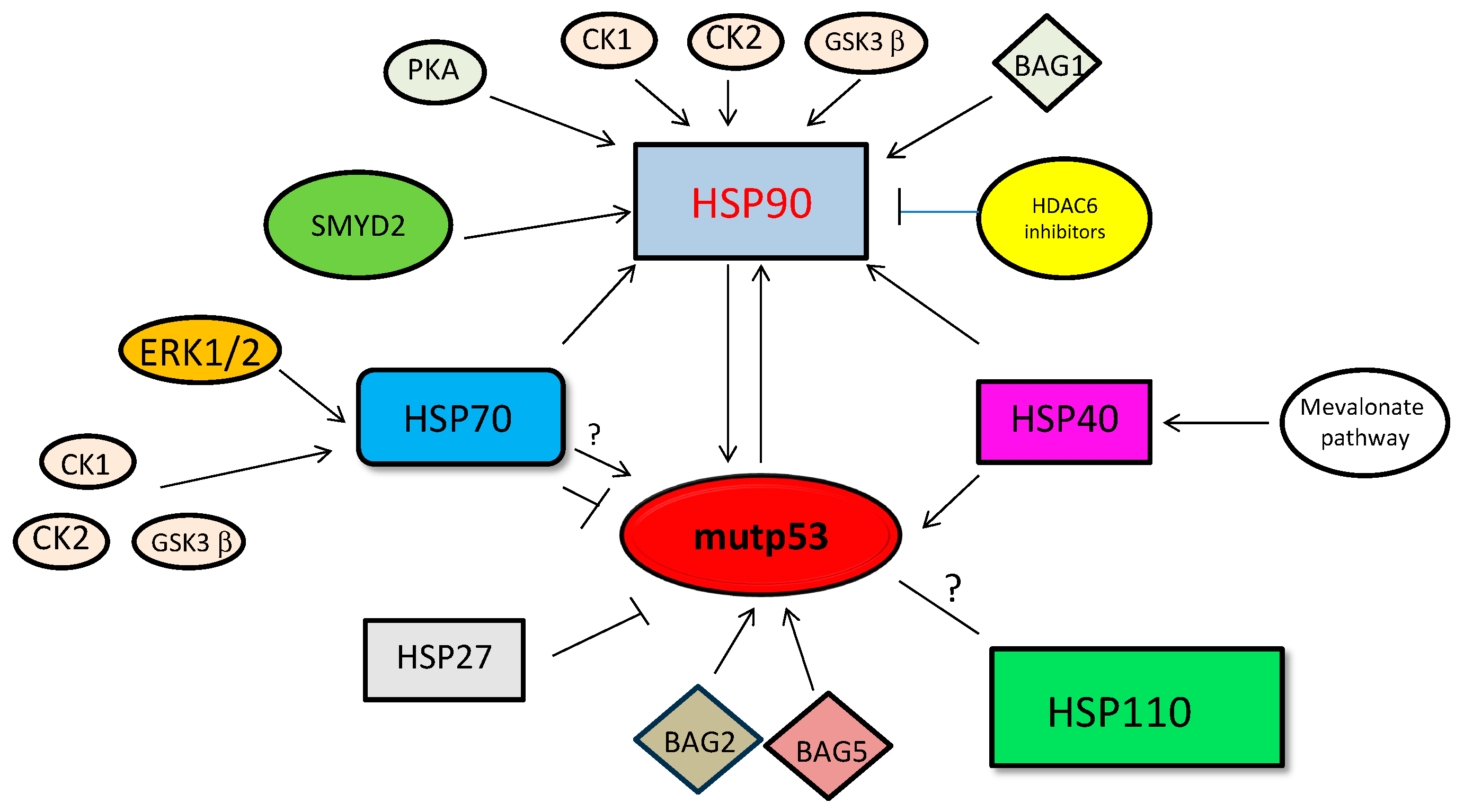
3.4. Targeting HSP Expression and Posttranslational Modifications to Destabilize Mutp53
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar] [PubMed]
- Resnick, M.A.; Inga, A. Functional mutants of the sequence-specific transcription factor p53 and implications for master genes of diversity. Proc. Natl. Acad. Sci. USA 2003, 100, 9934–9939. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.L.F.; Lee, M.Y.; Goh, H.C.; Ng, G.Y.N.; Lee, J.J.H.; Kannan, S.; Lim, Y.T.; Zhao, T.; Lim, E.K.H.; Phua, C.Z.J.; et al. Domain-specific p53 mutants activate EGFR by distinct mechanisms exposing tissue-independent therapeutic vulnerabilities. Nat. Commun. 2023, 14, 1726. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xie, T.; Wang, B.; Wang, R.; Cai, Y.; Yuan, B.; Gleber-Netto, O.F.; Tian, X.; Rodriguez-Rosario, A.E.; Osman, A.A.; et al. Mutant p53 drives an immune cold tumor immune microenvironment in oral squamous cell carcinoma. Commun. Biol. 2022, 5, 757. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Santos, G.S.; Zhan, Y.; Oliveira, M.M.S.; Rezaei, S.; Singh, M.; Peuget, S.; Westerberg, L.S.; Johnsen, J.I.; Selivanova, G. Mutant p53 gain of function mediates cancer immune escape that is counteracted by APR-246. Br. J. Cancer 2022, 127, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Lonetto, G.; Koifman, G.; Silberman, A.; Attery, A.; Solomon, H.; Levin-Zaidman, S.; Goldfinger, N.; Porat, Z.; Erez, A.; Rotter, V. Mutant p53-dependent mitochondrial metabolic alterations in a mesenchymal stem cell-based model of progressive malignancy. Cell Death Differ. 2019, 26, 1566–1581. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, E.; Takacs, E.M.; Kaur, S.; Cheng, C.; Kurokawa, M. Predicting clinical outcomes of cancer patients with a p53 deficiency gene signature. Sci. Rep. 2022, 12, 1317. [Google Scholar] [CrossRef] [PubMed]
- Bian, C.; Li, Z.; Xu, Y.; Wang, J.; Xu, L.; Shen, H. Clinical outcome and expression of mutant P53, P16, and Smad4 in lung adenocarcinoma: A prospective study. World J. Surg. Oncol. 2015, 13, 128. [Google Scholar] [CrossRef]
- Choundhury, S.; Kolukula, V.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Kaida, A.; Iwakuma, T. Regulation of p53 and Cancer Signaling by Heat Shock Protein 40/J-Domain Protein Family Members. Int. J. Mol. Sci. 2021, 22, 13527. [Google Scholar] [CrossRef] [PubMed]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1α/UCP2 axis increasing mitochondrial O2-· production in cancer cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Cordani, M.; Donadelli, M. Mutant p53 and mTOR/PKM2 regulation in cancer cells. IUBMB Life 2016, 68, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Schofield, H.K.; Zeller, J.; Espinoza, C.; Halbrook, C.J.; del Vecchio, A.; Magnuson, B.; Fabo, T.; Cali Daylan, A.E.; Kovalenko, I.; Lee, H.J.; et al. Mutant p53R270H drives altered metabolism and increased invasion in pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, e97422. [Google Scholar] [CrossRef] [PubMed]
- Kollareddy, M.; Dimitrova, E.; Vallabhaneni, K.C.; Chan, A.; Le, T.; Chauhan, K.M.; Carrero, Z.I.; Ramakrishnan, G.; Watabe, K.; Haupt, Y.; et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat. Commun. 2015, 6, 7389. [Google Scholar] [CrossRef]
- Tombari, C.; Zannini, A.; Bertolio, R.; Pedretti, S.; Audano, M.; Triboli, L.; Cancila, V.; Vacca, D.; Caputo, M.; Donzelli, S.; et al. Mutant p53 sustains serine-glycine synthesis and essential amino acids intake promoting breast cancer growth. Nat. Commun. 2023, 14, 6777. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Blee, A.M.; Zhang, J.; Pan, Y.; Becker, N.A.; Maher, L.J.; Jimenez, R.; Wang, L.; Huang, H. Gain-of-function mutant p53 together with ERG proto-oncogene drive prostate cancer by beta-catenin activation and pyrimidine synthesis. Nat. Commun. 2023, 14, 4671. [Google Scholar] [CrossRef]
- Wang, Z.H.; Peng, W.B.; Zhang, P.; Yang, X.P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell Biol. 2017, 37, e00328-17. [Google Scholar] [CrossRef] [PubMed]
- Chaudagar, K.; Hieromnimon, H.M.; Khurana, R.; Labadie, B.; Hirz, T.; Mei, S.; Hasan, R.; Shafran, J.; Kelley, A.; Apostolov, E.; et al. Reversal of Lactate and PD-1–mediated Macrophage Immunosuppression Controls Growth of PTEN/p53-deficient Prostate Cancer. Clin. Cancer Res. 2023, 29, 1952–1968. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Pacchiana, R.; Butera, G.; D’Orazi, G.; Scarpa, A.; Donadelli, M. Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: Involvement in cancer invasion and metastasis. Cancer Lett. 2016, 376, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Butera, G.; Brandi, J.; Cavallini, C.; Scarpa, A.; Lawlor, R.T.; Scupoli, M.T.; Marengo, E.; Cecconi, D.; Manfredi, M.; Donadelli, M. The Mutant p53-Driven Secretome Has Oncogenic Functions in Pancreatic Ductal Adenocarcinoma Cells. Biomolecules 2020, 10, 884. [Google Scholar] [CrossRef] [PubMed]
- Capaci, V.; Bascetta, L.; Fantuz, M.; Beznoussenko, G.V.; Sommaggio, R.; Cancila, V.; Bisso, A.; Campaner, E.; Mironov, A.A.; Wiśniewski, J.R.; et al. Mutant p53 induces Golgi tubulo-vesiculation driving a prometastatic secretome. Nat. Commun. 2020, 11, 3945. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers 2023, 15, 1899. [Google Scholar] [CrossRef] [PubMed]
- O’Loghlen, A. Role for extracellular vesicles in the tumour microenvironment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160488. [Google Scholar] [CrossRef]
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; MacFarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 5069. [Google Scholar] [CrossRef]
- Bhatta, B.; Luz, I.; Krueger, C.; Teo, F.X.; Lane, D.P.; Sabapathy, K.; Cooks, T. Cancer Cells Shuttle Extracellular Vesicles Containing Oncogenic Mutant p53 Proteins to the Tumor Microenvironment. Cancers 2021, 13, 2985. [Google Scholar] [CrossRef]
- Ju, Q.; Zhao, L.; Gao, J.; Zhou, L.; Xu, Y.; Sun, Y.; Zhao, X. Mutant p53 increases exosome-mediated transfer of miR-21-3p and miR-769-3p to promote pulmonary metastasis. Chin. J. Cancer Res. 2019, 31, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; McGuire, M.H.; Mangala, L.S.; Lee, S.; Stur, E.; Hu, W.; Bayraktar, E.; Villar-Prados, A.; Ivan, C.; Wu, S.Y.; et al. Gain-of-function p53 protein transferred via small extracellular vesicles promotes conversion of fibroblasts to a cancer-associated phenotype. Cell Rep. 2021, 34, 108726. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, C.; Deng, C.; Liu, J.; Li, D.; Zhou, T.; Yang, X.; Liu, Y.; Guo, Q.; Feng, Y.; et al. Regulated secretion of mutant p53 negatively affects T lymphocytes in the tumor microenvironment. Oncogene 2024, 43, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Cordani, M.; Zarrabi, A.; Alcon-Rodriguez, S.; Sainz, R.M.; Velasco, G.; Gonzalez-Menendez, P.; Dando, I. Transcending frontiers in prostate cancer: The role of oncometabolites on epigenetic regulation, CSCs, and tumor microenvironment to identify new therapeutic strategies. Cell Commun. Signal. 2024, 22, 36. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yu, J.; Li, F.; Ge, S. Oncometabolites drive tumorigenesis by enhancing protein acylation: From chromosomal remodelling to nonhistone modification. J. Exp. Clin. Cancer Res. 2022, 41, 144. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, T.; Ortega-Bernal, D.; Herrera, L.A.; González-De la Rosa, C.H.; Domínguez-Gómez, G.; Aréchaga-Ocampo, E.; Díaz-Chávez, J. Mutant p53 Gain-of-Function Induces Migration and Invasion through Overexpression of miR-182-5p in Cancer Cells. Cells 2023, 12, 2506. [Google Scholar] [CrossRef] [PubMed]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function p53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Santarelli, R.; D’Orazi, G.; Cirone, M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front. Oncol. 2020, 10, 2985. [Google Scholar] [CrossRef]
- Lakoduk, A.M.; Roudot, P.; Mettlen, M.; Grossman, H.M.; Schmid, S.L.; Chen, P.H. Mutant p53 amplifies a dynamin-1/APPL1 endosome feedback loop that regulates recycling and migration. J. Cell Biol. 2019, 218, 1928–1942. [Google Scholar] [CrossRef]
- Mukherjee, S.; Maddalena, M.; Lü, Y.; Martinez, S.; Nataraj, N.B.; Noronha, A.; Sinha, S.; Teng, K.; Cohen-Kaplan, V.; Ziv, T.; et al. Cross-talk between mutant p53 and p62/SQSTM1 augments cancer cell migration by promoting the degradation of cell adhesion proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2119644119. [Google Scholar] [CrossRef]
- Pavlakis, E.; Neumann, M.; Merle, N.; Wieboldt, R.; Wanzel, M.; Ponath, V.; Pogge von Strandmann, E.; Elmshäuser, S.; Stiewe, T. Mutant p53-ENTPD5 control of the calnexin/calreticulin cycle: A druggable target for inhibiting integrin-α5-driven metastasis. J. Exp. Clin. Cancer Res. 2023, 42, 203. [Google Scholar] [CrossRef]
- Sun, S.; Chen, H.; Sun, L.; Wang, M.; Wu, X.; Xiao, Z.X.J. Hotspot mutant p53-R273H inhibits KLF6 expression to promote cell migration and tumor metastasis. Cell Death Dis. 2020, 11, 595. [Google Scholar] [CrossRef]
- Lv, T.; Lv, H.; Fei, J.; Xie, Y.; Lian, D.; Hu, J.; Tang, L.; Shi, X.; Wang, J.; Zhang, S.; et al. p53-R273H promotes cancer cell migration via upregulation of neuraminidase-1. J. Cancer 2020, 11, 6874–6882. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Cordani, M.; Mullappilly, N.; Pacchiana, R.; Riganti, C.; Palmieri, M.; Pons, D.G.; Roca, P.; Oliver, J.; Donadelli, M. Mutant p53 induces SIRT3/MnSOD axis to moderate ROS production in melanoma cells. Arch. Biochem. Biophys. 2020, 679, 108219. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Beaver, J.M.; Liu, Y.; Zhang, Z. Dynamics of p53: A Master Decider of Cell Fate. Genes 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Stanga, S.; Lanni, C.; Govoni, S.; Uberti, D.; D’Orazi, G.; Racchi, M. Unfolded p53 in the pathogenesis of Alzheimer’s disease: Is HIPK2 the link? Aging 2010, 2, 545–554. [Google Scholar] [CrossRef]
- Kupis, W.; Pałyga, J.; Tomal, E.; Niewiadomska, E. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Starace, G.; Rechavi, G.; Sacchi, A.; Givol, D.; D’Orazi, G. Nox1 is involved in p53 deacetylation and suppression of its transcriptional activity and apoptosis. Free Radic. Biol. Med. 2010, 48, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Aventaggiato, M.; Vernucci, E.; Barreca, F.; Russo, M.A.; Tafani, M. Sirtuins’ control of autophagy and mitophagy in cancer. Pharmacol. Ther. 2021, 221, 107748. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhou, L.M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.Y.; Lu, X.T.; Hou, M.L.; Cao, T.; Tian, Z. Sirtuin1-p53: A potential axis for cancer therapy. Biochem. Pharmacol. 2023, 212, 115543. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef]
- Perez, R.E.; Knights, C.D.; Sahu, G.; Catania, J.; Kolukula, V.K.; Stoler, D.; Graessmann, A.; Ogryzko, V.; Pishvaian, M.; Albanese, C.; et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J. Cell Physiol. 2010, 225, 394–405. [Google Scholar] [CrossRef]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, T.; Buschmann, T.; Habelhah, H.; Matusevich, E.; Tahara, H.; Boerresen-Dale, A.L.; Harris, C.; Sidransky, D.; Ronai, Z. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001, 20, 3341–3347. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary downregulation of mutant p53 levels via glucose restriction: Mechanisms and implications for tumor therapy. Cell Cycle 2012, 11, 4436–4446. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, Y.; Liu, L.; Guo, R.; Zhang, P.; Zhang, Y.; Zhang, Y.; Zhao, J.; Su, J.; Sun, L.; et al. Sirtuin 3 induces apoptosis and necroptosis by regulating mutant p53 expression in small-cell lung cancer. Oncol. Rep. 2020, 43, 591–600. [Google Scholar] [CrossRef]
- Tao, R.; Vassilopoulos, A.; Parisiadou, L.; Yan, Y.; Gius, D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid. Redox Signal. 2014, 20, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Treviño-Saldaña, N.; García-Rivas, G. Regulation of Sirtuin-Mediated Protein Deacetylation by Cardioprotective Phytochemicals. Oxid. Med. Cell Longev. 2017, 2017, 1750306. [Google Scholar] [CrossRef]
- Suenkel, B.; Valente, S.; Zwergel, C.; Weiss, S.; Di Bello, E.; Fioravanti, R.; Aventaggiato, M.; Amorim, J.A.; Garg, N.; Kumar, S.; et al. Potent and Specific Activators for Mitochondrial Sirtuins Sirt3 and Sirt5. J. Med. Chem. 2022, 65, 14015–14031. [Google Scholar] [CrossRef] [PubMed]
- Zwergel, C.; Aventaggiato, M.; Garbo, S.; Di Bello, E.; Fassari, B.; Noce, B.; Castiello, C.; Lambona, C.; Barreca, F.; Rotili, D.; et al. Novel 1,4-Dihydropyridines as Specific Binders and Activators of SIRT3 Impair Cell Viability and Clonogenicity and Downregulate Hypoxia-Induced Targets in Cancer Cells. J. Med. Chem. 2023, 66, 9622–9641. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.J.; To, T.S.S.; Zhao, H.F.; Wang, J. Role of SIRT1-mediated mitochondrial and Akt pathways in glioblastoma cell death induced by Cotinus coggygria flavonoid nanoliposomes. Int. J. Nanomed. 2015, 10, 5005–5023. [Google Scholar]
- Wang, G.; Wang, J.J.; Wang, Y.Z.; Feng, S.; Jing, G.; Fu, X.L. Myricetin nanoliposomes induced SIRT3-mediated glycolytic metabolism leading to glioblastoma cell death. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. S3), S180–S191. [Google Scholar] [CrossRef] [PubMed]
- Aventaggiato, M.; Barreca, F.; Sansone, L.; Pellegrini, L.; Russo, M.A.; Cordani, M.; Tafani, M. Sirtuins and Hypoxia in EMT Control. Pharmaceuticals 2022, 15, 737. [Google Scholar] [CrossRef] [PubMed]
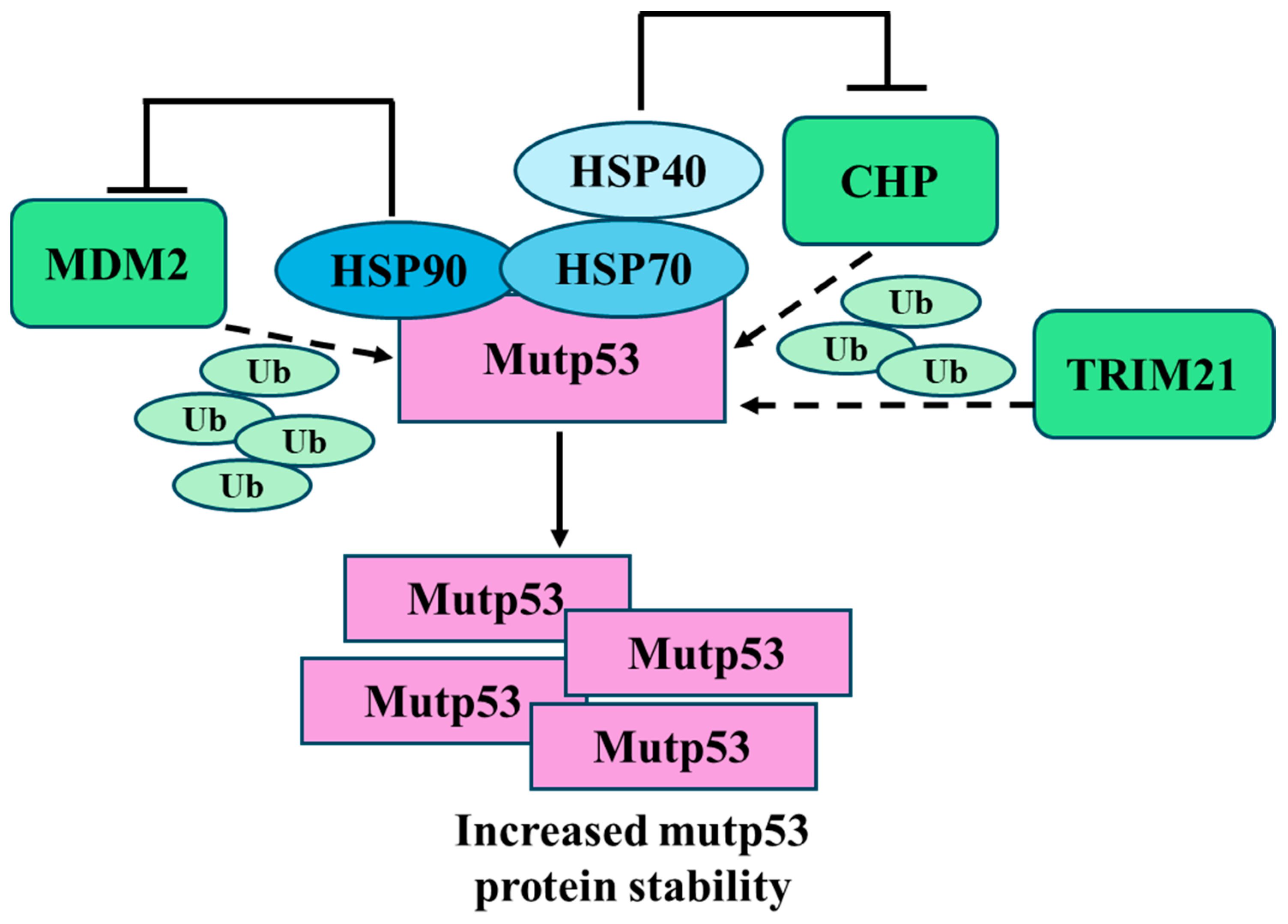
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol.Cell Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, W.; Zhang, L.; Zhang, J. Targeting mutant p53 stabilization for cancer therapy. Front. Pharmacol. 2023, 14, 1215995. [Google Scholar] [CrossRef] [PubMed]
- do Patrocinio, A.B.; Rodrigues, V.; Guidi Magalhães, L. P53: Stability from the Ubiquitin–Proteasome System and Specific 26S Proteasome Inhibitors. ACS Omega 2022, 7, 3836–3843. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Gu, W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010, 17, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16 INK4a loss. Genes. Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; White, E. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev. 2008, 22, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, S.; Li, Q.; Hu, S.; Tashakori, M.; Van Pelt, C.; You, M.J.; Pageon, L.; Lozano, G. Tissue-specific and age-dependent effects of global Mdm2 loss. J. Pathol. 2014, 233, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Romeo, M.A.; Gilardini Montani, M.S.; Arena, A.; Benedetti, R.; D’Orazi, G.; Cirone, M. c-Myc Sustains Pancreatic Cancer Cell Survival and mutp53 Stability through the Mevalonate Pathway. Biomedicines 2022, 10, 2489. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Xu, D.; Zhang, T.; Chang, C.Y.; Wang, J.; Liu, J.; Zhang, L.; Haffty, B.G.; Zong, W.X.; et al. The ubiquitin ligase TRIM21 regulates mutant p53 accumulation and gain of function in cancer. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Shi, Y.; Shen, H.M.; Gopalakrishnan, V.; Gordon, N. Epigenetic Regulation of Autophagy Beyond the Cytoplasm: A Review. Front. Cell Dev. Biol. 2021, 9, 675599. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays. 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-Mediated Autophagy. 2008. Available online: https://pubmed.ncbi.nlm.nih.gov/18425454/ (accessed on 6 May 2024).
- LaGory, E.L.; Giaccia, A.J. A low-carb diet kills tumor cells with a mutant p53 tumor suppressor gene. Cell Cycle 2013, 12, 718–719. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reactivation of mutant p53 by capsaicin, the major constituent of peppers. J. Exp. Clin. Cancer Res. 2016, 35, 136. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Federici, G.; Gilardini Montani, M.S.; Crispini, A.; Cirone, M.; D’Orazi, G. Interplay between Endoplasmic Reticulum (ER) Stress and Autophagy Induces Mutant p53H273 Degradation. Biomolecules 2020, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef] [PubMed]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Monti, P.; Degan, P.; Fronza, G.; Menichini, P. Gambogic acid counteracts mutant p53 stability by inducing autophagy. Biochim. Et. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Baldari, S.; Ubertini, V.; Garufi, A.; D’Orazi, G.; Bossi, G. Targeting MKK3 as a novel anticancer strategy: Molecular mechanisms and therapeutical implications. Cell Death Dis. 2015, 6, e1621. [Google Scholar] [CrossRef]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Trisciuoglio, D.; Porru, M.; Leonetti, C.; Stoppacciaro, A.; D’Orazi, V.; Avantaggiati, M.; Crispini, A.; Pucci, D.; D’Orazi, G. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R175H and R273H) p53 in cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 72. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pistritto, G.; Baldari, S.; Toietta, G.; Cirone, M.; D’Orazi, G. p53-Dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions. J. Exp. Clin. Cancer Res. 2017, 36, 126. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. Cell Physiol. 1995, 269, C1200-8. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes. Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Galan-Acosta, L.; Norberg, E. Glucose metabolism provide distinct prosurvival benefits to non-small cell lung carcinomas. Biochem. Biophys. Res. Commun. 2015, 460, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Romeo, M.A.; Arena, A.; Gilardini Montani, M.S.; D’Orazi, G.; Cirone, M. ATF6 supports lysosomal function in tumor cells to enable ER stress-activated macroautophagy and CMA: Impact on mutant TP53 expression. Autophagy 2024, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Jointed by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Kim, J.; Kim, K.I. Crosstalk between endoplasmic reticulum stress response and autophagy in human diseases. Anim. Cells Syst. 2023, 27, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pistritto, G.; D’Orazi, V.; Cirone, M.; D’Orazi, G. The Impact of NRF2 Inhibition on Drug-Induced Colon Cancer Cell Death and p53 Activity: A Pilot Study. Biomolecules 2022, 12, 461. [Google Scholar] [CrossRef]
- Russell, R.C.; Guan, K. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Cordani, M.; Donadelli, M.; Ghavami, S. Metastatic outgrowth via the two-way interplay of autophagy and metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1870, 166824. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, L.; Bodemeyer, V.; Bosisio, F.M.; Claps, G.; Carretta, M.; Rizza, S.; Faienza, F.; Frias, A.; Khan, S.; Bordi, M.; et al. Loss of Ambra1 promotes melanoma growth and invasion. Nat. Commun. 2021, 12, 2550. [Google Scholar] [CrossRef]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-Function Mutant p53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Crown, J. Drugging ‘undruggable’ genes for cancer treatment: Are we making progress? Int. J. Cancer 2021, 148, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm 2022, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol. Cell 2019, 74, 831–843.e4. [Google Scholar] [CrossRef] [PubMed]
- Helmbrecht, K.; Zeise, E.; Rensing, L. Chaperones in cell cycle regulation and mitogenic signal transduction: A review. Cell Prolif. 2000, 33, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar] [CrossRef]
- Muller, P.; Hrstka, R.; Coomber, D.; Lane, D.P.; Vojtesek, B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008, 27, 3371–3383. [Google Scholar] [CrossRef]
- Li, D.; Yallowitz, A.; Ozog, L.; Marchenko, N. A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis. 2014, 5, e1194. [Google Scholar] [CrossRef]
- Zylicz, M.; King, F.W.; Wawrzynow, A. Hsp70 interactions with the p53 tumour suppressor protein. EMBO J. 2001, 20, 4634–4638. [Google Scholar] [CrossRef]
- Garufi, A.; Baldari, S.; Pettinari, R.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Crispini, A.; Giorno, E.; Toietta, G.; Marchetti, F.; et al. A ruthenium(II)-curcumin compound modulates NRF2 expression balancing the cancer cell death/survival outcome according to p53 status. J. Exp. Clin. Cancer Res. 2020, 39, 122. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A. Cirone M Mutant p53, Stabilized by Its Interplay with HSP90, Activates a Positive Feed-Back Loop between NRF2 and p62 that Induces Chemo-Resistance to Apigenin in Pancreatic Cancer Cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Arena, A.; D’Orazi, G.; Cirone, M. VPA and TSA Interrupt the Interplay between mutp53 and HSP70, Leading to CHK1 and RAD51 Down-Regulation and Sensitizing Pancreatic Cancer Cells to AZD2461 PARP Inhibitor. Int. J. Mol. Sci. 2022, 23, 2268. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.; Bosco, B.; Loureiro, J.B.; Ramos, H.; Raimundo, L.; Soares, J.; Nazareth, N.; Barcherini, V.; Domingues, L.; Oliveira, C.; et al. SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers 2019, 11, 1151. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- King, F.W.; Wawrzynow, A.; Höhfeld, J.; Zylicz, M. Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 2001, 20, 6297–6305. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Scheffner, M.; Höhfeld, J. The Chaperone-associated Ubiquitin Ligase CHIP Is Able to Target p53 for Proteasomal Degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Gallardo, L.; Martín-Benito, J.; Marcilla, M.; Espadas, G.; Sabidó, E.; Valpuesta, J.M. The cochaperone CHIP marks Hsp70- and Hsp90-bound substrates for degradation through a very flexible mechanism. Sci. Rep. 2019, 9, 5102. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Hwang, S.Y.; Cao, S.; Ramadhar, T.R.; Byun, S.; Yoon, K.W.; Lee, J.H.; Chu, K.; Gurkar, A.U.; Kolev, V.; et al. Small-Molecule Reactivation of Mutant p53 to Wild-Type-like p53 through the p53-Hsp40 Regulatory Axis. Chem. Biol. 2015, 22, 1206–1216. [Google Scholar] [CrossRef]
- Nishikawa, S.; Kaida, A.; Parrales, A.; Ranjan, A.; Alalem, M.; Ren, H.; Schoenen, F.J.; Johnson, D.K.; Iwakuma, T. DNAJA1- and conformational mutant p53-dependent inhibition of cancer cell migration by a novel compound identified through a virtual screen. Cell Death Discov. 2022, 8, 437. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan-Sunol, C.; Gabai, V.L.; Sherman, M.Y. Hsp27 Modulates p53 Signaling and Suppresses Cellular Senescence. Cancer Res. 2007, 67, 11779–11788. [Google Scholar] [CrossRef] [PubMed]
- Kanagasabai, R.; Krishnamurthy, K.; Druhan, L.J.; Ilangovan, G. Forced Expression of Heat Shock Protein 27 (Hsp27) Reverses P-Glycoprotein (ABCB1)-mediated Drug Efflux and MDR1 Gene Expression in Adriamycin-resistant Human Breast Cancer Cells. J. Biol. Chem. 2011, 286, 33289–33300. [Google Scholar] [CrossRef] [PubMed]
- Nitika Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, D.G.; Kim, S. ERK-dependent phosphorylation of the linker and substrate-binding domain of HSP70 increases folding activity and cell proliferation. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef]
- Barra, J.; Cerda-Infante, J.; Sandoval, L.; Gajardo-Meneses, P.; Henriquez, J.F.; Labarca, M.; Metz, C.; Venegas, J.; Retamal, C.; Oyanadel, C.; et al. D-Propranolol Impairs EGFR Trafficking and Destabilizes Mutant p53 Counteracting AKT Signaling and Tumor Malignancy. Cancers 2021, 13, 3622. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Park, J.H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.J.; Lee, H.S.; Jang, S.H.; et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.Y.; Zhang, X.H.; Zhao, M.; Hou, C.M.; Xu, Y.J.; Du, Z.Y.; Yu, X.D. FK228 inhibits Hsp90 chaperone function in K562 cells via hyperacetylation of Hsp70. Biochem. Biophys. Res. Commun. 2007, 356, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Zahn, A.; Methot, S.P.; Godin, D.; Conticello, S.G.; Terada, K.; Di Noia, J.M. Optimal functional levels of activation-induced deaminase specifically require the Hsp40 DnaJa1. EMBO J. 2012, 31, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.E.; Moen, A.; Bousset, L.; Egge-Jacobsen, W.; Kernstock, S.; Melki, R.; Falnes, P.Ø. Identification and Characterization of a Novel Human Methyltransferase Modulating Hsp70 Protein Function through Lysine Methylation. J. Biol. Chem. 2013, 288, 27752–27763. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, R.; Toyokawa, G.; Nakakido, M.; Ueda, K.; Nakamura, Y. SMYD2-dependent HSP90 methylation promotes cancer cell proliferation by regulating the chaperone complex formation. Cancer Lett. 2014, 351, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Han, T.S.; Hur, K.; Cho, H.S. Epigenetic Alterations of Heat Shock Proteins (HSPs) in Cancer. Int. J. Mol. Sci. 2019, 20, 4758. [Google Scholar] [CrossRef] [PubMed]
- Coban, N.; Varol, N. The effect of heat shock protein 90 inhibitors on histone 4 lysine 20 methylation in bladder cancer. EXCLI J. 2019, 18, 195–203. [Google Scholar]
- Tan, Y.S.; Mhoumadi, Y.; Verma, C.S. Roles of computational modelling in understanding p53 structure, biology, and its therapeutic targeting. J. Mol. Cell Biol. 2019, 11, 306–316. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant p53 Proteins | Role in Cancer | Biological Effect Led by Mutant p53 | References |
|---|---|---|---|
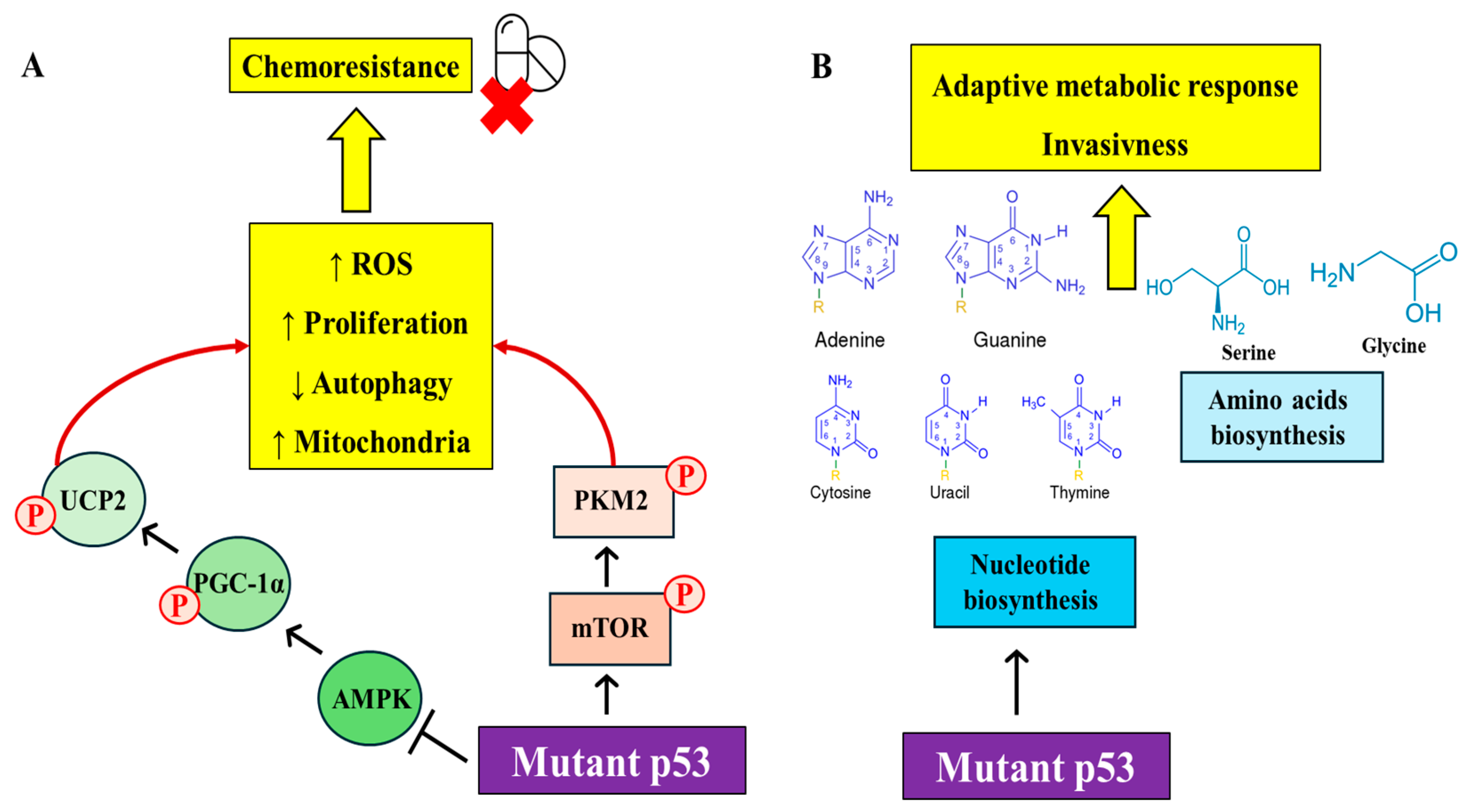
| p53R175H, p53R273H | Induces changes enhancing cancer aggressiveness | Triggers metabolic alterations increasing mitochondrial oxidation | [8] |
| p53R175H, p53R273H | Enhances pro-oxidant state and drug resistance | Inhibits the axis leading to hyper-proliferation, anti-apoptotic effects, and drug resistance | [14] |
| p53R273H, p53R280K | Enhances glycolysis and ROS production | Phosphorylation of PKM2 via mTOR signaling, reduces autophagy, supports oncogenic activity | [15] |
| p53R273H | Alters metabolism promoting EMT and invasion | Essential for precursor lesions, mediates malignant potential through metabolic changes | [16] |
| p53R248Q, p53R273H | Enhances invasiveness and cancer development | Associates with promoters of nucleotide metabolism genes, enhances GTP-dependent processes | [17] |
| p53R248Q, p53R273H | Stimulates synthesis crucial for survival under nutrient limitations | Reprograms metabolism, particularly serine/glycine synthesis, crucial in breast cancer | [18] |
| p53R273H, p53R280K | Accelerates oncogenesis | Transactivates genes involved in pyrimidine synthesis, associated with TMPRSS2-ERG fusion in prostate cancer | [19] |
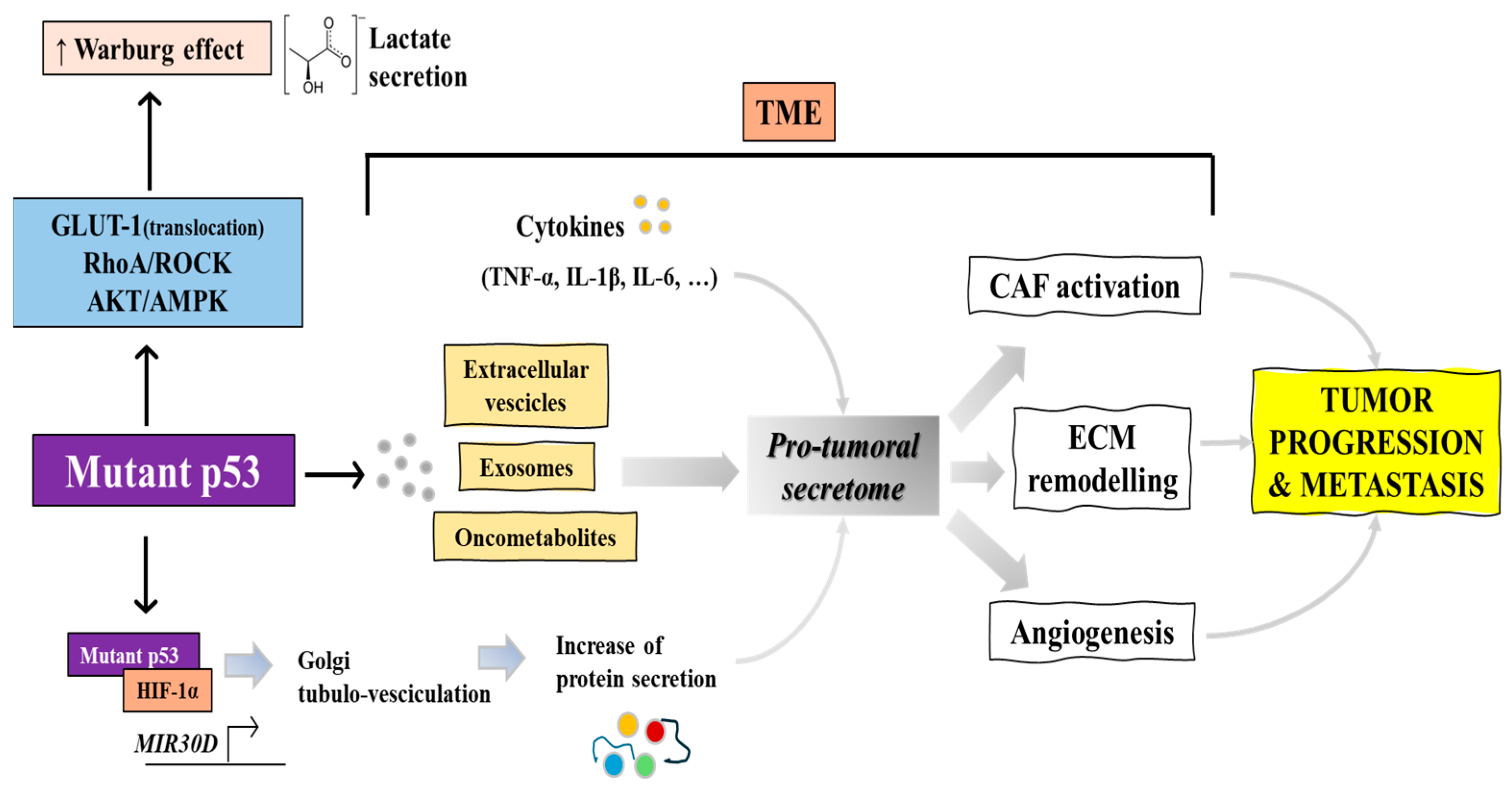
| p53R175H, p53R248Q, p53R273H | Stimulates enhanced glycolysis and lactate production | Promotes GLUT-1 translocation, affects RhoA/ROCK and AKT/AMPK signaling, enhances cancer cell survival and growth | [21] |
| p53R175H, p53R248Q, p53R273H | Alters cellular metabolism | Influences both glycolytic and mitochondrial oxidative phosphorylation pathways, impacts metabolic diversity | [22] |
| p53R175H, p53R248Q, p53R273H | Influences the secretome impacting immune responses | Shift in metabolism influences the secretome of cancer cells, impacting tumor microenvironment and immune evasion | [23] |
| p53R175H, p53R273H | Supports invasive cancer phenotypes | Changes in secretion of enzymes, inflammatory cytokines (IL-6, IL-1β, TNFα), and factors increasing extracellular acidification | [24] |
| p53R175H, p53R273H | Affects cancer progression | Alters cell secretome impacting proliferation, chemoresistance, apoptosis, EMT | [25] |
| p53R175H, p53R273H | Enhances malignant factor release | Alters Golgi structure and function, mediated by mut-p53/HIF1α/miR-30d axis leading to increased pro-malignant factor release | [26] |
| p53R273H, others not specified | Enhances cancer invasiveness | Alters the composition and function of EVs, facilitating the transfer of invasive properties across the TME. | [29] |
| p53R273H | Modifies TME, supports tumor progression | Transferring gain-of-function properties to neighboring cells and macrophages, thus promoting tumor growth | [30] |
| p53R273H | Influences ECM remodeling and EMT | Alters the TME through exosomal-micro (mi)RNAs that impact fibroblast activation and cytokine secretion, promoting EMT and enhancing tumor-stroma communication. | [31] |
| p53Y234C, p53G245D, p53I195T, p53R248W, p53R273H, p53Y163C | Leads to cancer-associated phenotypes and enhances tumor growth | GOF p53 proteins are packaged into small EVs and transferred to fibroblasts, mediated by HSP90, activating pathways like Nrf2 that induce cancer-associated transformations | [32] |
| p53R273H | Suppresses immune responses, notably CD4+ T lymphocytes, and alters glycolysis | Mutp53 secretion via exosomes impacts the immune microenvironment | [33] |
| p53R273H, p53R249S | Reprograms macrophages into a tumor-supportive state | Cancer cells shed miR-1246-enriched exosomes, which reprogram macrophages, contributing to an immunosuppressive environment | [34] |
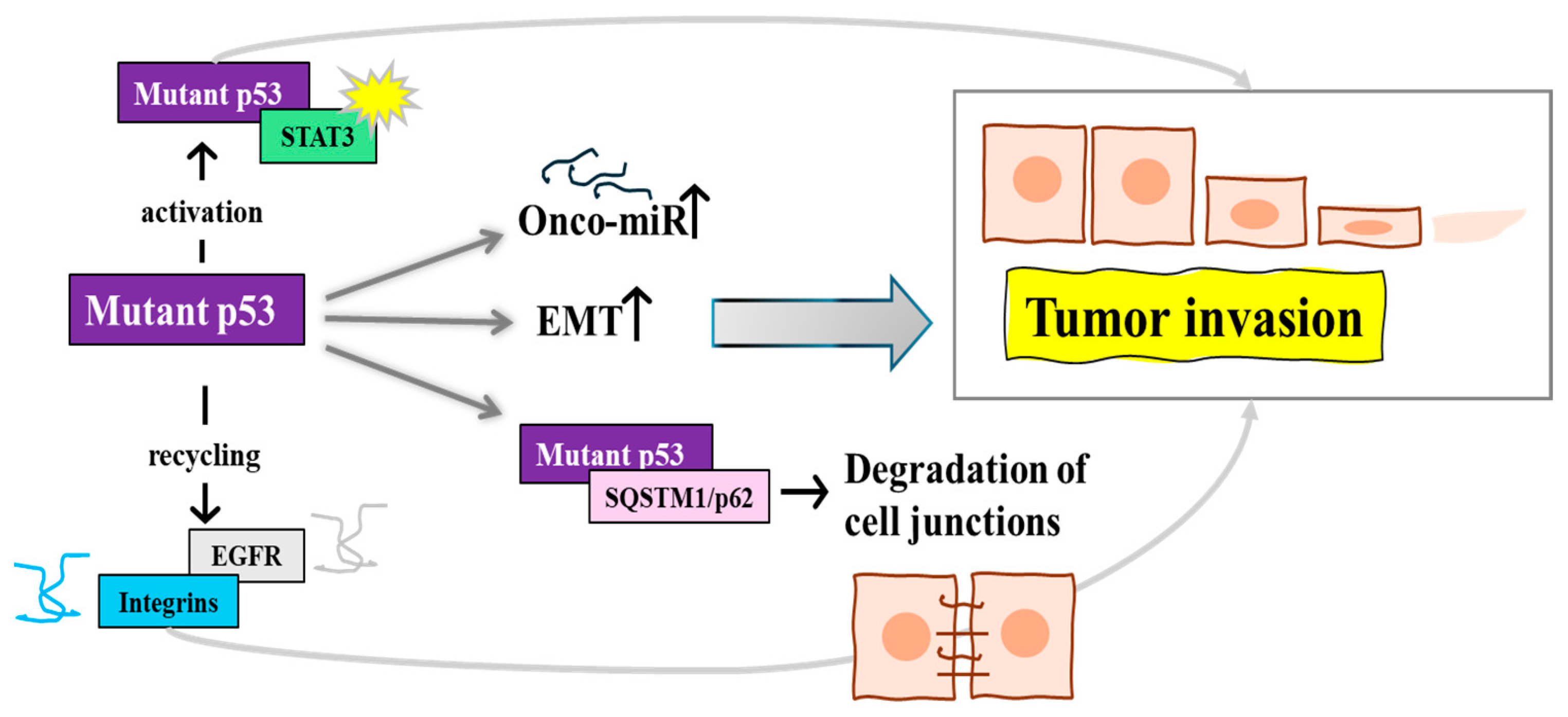
| General mutp53 (specific mutations not detailed) | Enhances cell migration and invasion | Modulates miRNA expression, such as upregulation of miR-182-5p, affecting gene regulation pathways related to migration and invasion | [37] |
| General mutp53 (in PDAC, colorectal cancers, glioblastoma) | Promotes tumor progression and metastasis | Binds to and hyperactivates STAT3, leading to enhanced tumor progression, invasion, and metastasis | [38,39] |
| p53R273H | Increases cell migration and invasion | Enhances rapid recycling of EGFR and β1 integrins, crucial for cancer cell motility | [40] |
| p53R273H | Facilitates cancer cell migration | Interacts with SQSTM1/p62 leading to the degradation of cell-junction-associated proteins | [41] |
| p53R273H, p53R248W, p53R248Q, p53R280K | Affects tumor cell motility | Influences the mutp53-ENTPD5 axis in N-glycoprotein biosynthesis, impacting tumor-stroma interactions and motility | [42] |
| p53R273H | Promotes cell migration and metastasis | Suppresses tumor suppressor genes like KLF6, impacting E-cadherin levels and promoting cell migration and metastasis | [43] |
| p53R273H | Enhances cell migration | Upregulates NEU1, a sialidase involved in cell migration, highlighting diverse mechanisms enhancing metastasis | [44] |
| p53R273H, p53E258K | Enhances tumor progression | Mutp53 induces MnSOD mRNA and protein expression, enhancing aerobic glycolysis and tumor progression through balanced ROS production and reduced oxidative damage | [45] |
| Mutant p53 Proteins | Agents/Compounds | References |
|---|---|---|
| p53R175H, p53R273H | curcumin-based zinc compound (Zn(II), curcumin compounds and capsaicin (8-methyl-N-vanillyl-6-noneamide) | [96,97,98] |
| p53R280K, p53S241F | Gambogic acid | [99] |
| p53R273H | inhibition of MKK3 reduces mutp53 levels through ER stress-induced autophagy | [100] |
| p53R175H, R273H, R248Q | cruciferous-vegetable-derived phenethyl isothiocyanate (PEITC), | [101] |
| p53R172H, R248Q, R280K | histone deacetylases inhibitors (HDACi) | [102] |
| P98S, P151H, A161T, R175C, R175D, R175H, L194F, S227K, S227R, G245C, R248L, R248W, E258K, R273H, R273L, R280K, R28W | Stimulation of CMA by macroautophagy inhibition or by inducing ER stress | [106,107,108] |
| R273H, R175H, R280K | HSP90 inhibition | [129] |
| p53R280K, p53R273K, p53L194F, p53R175H, p53P223L | HSP90, HSP70 acetylation | [152,153] |
| p53 R280K, p53 R273H, p53 M237I, p53 R249S, p53 R175H, p53 L194F p53 V157F, p53 R249S p53 R213Q | Mevalonate pathway inhibitors | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordani, M.; Garufi, A.; Benedetti, R.; Tafani, M.; Aventaggiato, M.; D’Orazi, G.; Cirone, M. Recent Advances on Mutant p53: Unveiling Novel Oncogenic Roles, Degradation Pathways, and Therapeutic Interventions. Biomolecules 2024, 14, 649. https://doi.org/10.3390/biom14060649
Cordani M, Garufi A, Benedetti R, Tafani M, Aventaggiato M, D’Orazi G, Cirone M. Recent Advances on Mutant p53: Unveiling Novel Oncogenic Roles, Degradation Pathways, and Therapeutic Interventions. Biomolecules. 2024; 14(6):649. https://doi.org/10.3390/biom14060649
Chicago/Turabian StyleCordani, Marco, Alessia Garufi, Rossella Benedetti, Marco Tafani, Michele Aventaggiato, Gabriella D’Orazi, and Mara Cirone. 2024. "Recent Advances on Mutant p53: Unveiling Novel Oncogenic Roles, Degradation Pathways, and Therapeutic Interventions" Biomolecules 14, no. 6: 649. https://doi.org/10.3390/biom14060649
APA StyleCordani, M., Garufi, A., Benedetti, R., Tafani, M., Aventaggiato, M., D’Orazi, G., & Cirone, M. (2024). Recent Advances on Mutant p53: Unveiling Novel Oncogenic Roles, Degradation Pathways, and Therapeutic Interventions. Biomolecules, 14(6), 649. https://doi.org/10.3390/biom14060649