
Advances in Integrated Multi-omics Analysis for Drug-Target Identification


Abstract
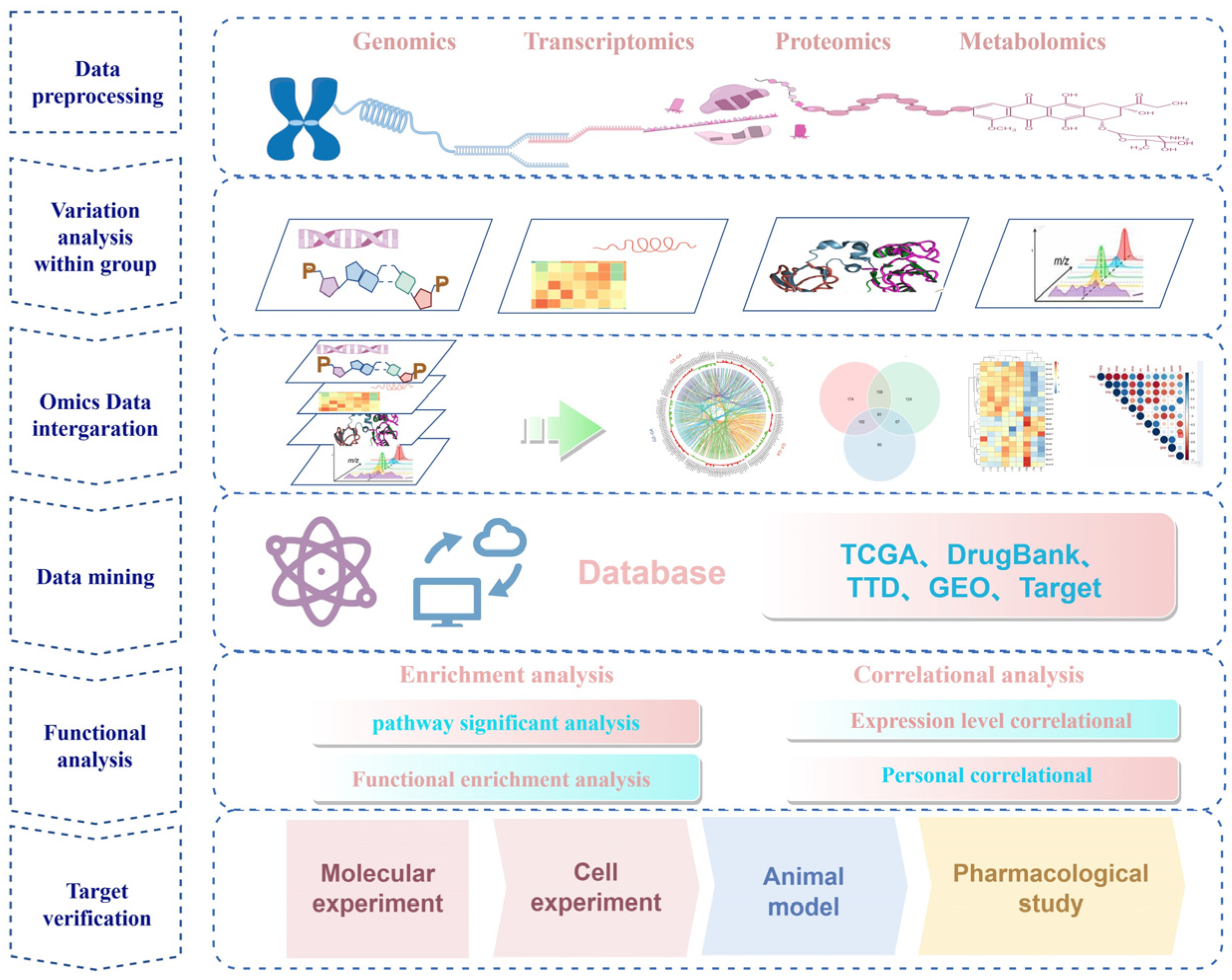
:1. Introduction
2. Development History of Omics Technologies
3. Single-Omics Analysis
3.1. Genomics
3.2. Transcriptomics
3.3. Proteomics
3.4. Metabolomics
4. Classic Multi-omics Techniques Analysis
5. Classical Multi-omics Integration Methods
5.1. Integrating Transcriptome and Proteomics
5.2. Integrating Transcriptome and Metabolomics
5.3. Integrating Proteomics and Metabolomics
5.4. Proteogenomics
6. Single-Cell Multi-omics Technology
7. Spatial Multi-omics Technology
8. Computational Tools for Multi-omics Data Integration
9. Future Perspective
9.1. Challenges
9.1.1. The Complexity of Data Integration and Analysis
9.1.2. Data Consistency and Standardization
9.1.3. The Rising Cost of Data Analysis
9.2. Prospects for Technology Application
9.3. Technological Advances and Breakthroughs
9.3.1. Single-Cell Multi-omics and Spatial Multi-omics
9.3.2. Multi-omics and Machine Learning
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-H.; Mao, N.-D.; Duan, J.-L.; Zhang, H.; Garrido, C.; Lirussi, F.; Gao, Y.; Xie, T.; Ye, X.-Y. Recent progress in discovery of novel AAK1 inhibitors: From pain therapy to potential anti-viral agents. J. Enzym. Inhib. Med. Chem. 2023, 38, 2279906. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Jiang, X.; Zhao, R.; Zhong, Z.; Ge, J.; Zhu, J.; Ye, X.-Y.; Xie, Y.; Liu, Z.; Xie, T. HDAC1/MAO-B dual inhibitors against Alzheimer’s disease: Design, synthesis and biological evaluation of N-propargylamine-hydroxamic acid/o-aminobenzamide hybrids. Bioorganic Chem. 2022, 122, 105724. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, H.; Zhang, Y.; Ye, Y.; Wang, S.; Bai, R.; Xie, T.; Ye, X.-Y. Drug discovery of histone lysine demethylases (KDMs) inhibitors (progress from 2018 to present). Eur. J. Med. Chem. 2022, 231, 114143. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.-L.; Wang, C.-C.; Yuan, Y.; Hui, Z.; Zhang, H.; Mao, N.-D.; Zhang, P.; Sun, B.; Lin, J.; Zhang, Z. Design, Synthesis, and Structure–Activity Relationship of Novel Pyridazinone-Based PARP7/HDACs Dual Inhibitors for Elucidating the Relationship between Antitumor Immunity and HDACs Inhibition. J. Med. Chem. 2024, 67, 4950–4976. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Ozerov, I.V.; Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 2023, 44, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Nim, S.; Teyra, J.; Datti, A.; Wrana, J.L.; Sidhu, S.S.; Moffat, J.; Kim, P.M. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 2014, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology; Wiley Online Library: Hoboken, NJ, USA, 2016; Volume 11, pp. 1190–1192. [Google Scholar]
- Pinheiro-de-Sousa, I.; Fonseca-Alaniz, M.H.; Giudice, G.; Valadão, I.C.; Modestia, S.M.; Mattioli, S.V.; Junior, R.R.; Zalmas, L.P.; Fang, Y.; Petsalaki, E. Integrated systems biology approach identifies gene targets for endothelial dysfunction. Mol. Syst. Biol. 2023, 19, e11462. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Sussulini, A.; Xia, J.; Orešič, M. Multi-omics: Trends and applications in clinical research. Front. Mol. Biosci. 2022, 9, 994239. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.V.; Hu, Y.-J. Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases. Adv. Genet. 2016, 93, 147–190. [Google Scholar] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-T.; Lu, Y.; Wu, J.; Zhang, C.; Tan, H.-Y.; Bian, Z.-X.; Wang, N.; Feng, Y. CRISPR-Cas9 library screening approach for anti-cancer drug discovery: Overview and perspectives. Theranostics 2022, 12, 3329. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.; Roudnicky, F. Functional genomics for cancer drug target discovery. Cancer Cell 2020, 38, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kassner, M. In vitro high-throughput RNAi screening to accelerate the process of target identification and drug development. In High-Throughput RNAi Screening: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2016; pp. 137–149. [Google Scholar]
- Adams, R.; Steckel, M.; Nicke, B.; Pohlenz, H.-D. RNAi as a tool for target discovery in early pharmaceutical research. Pharm.-Int. J. Pharm. Sci. 2016, 71, 35–42. [Google Scholar]
- Zhang, Q.; Major, M.B.; Takanashi, S.; Camp, N.D.; Nishiya, N.; Peters, E.C.; Ginsberg, M.H.; Jian, X.; Randazzo, P.A.; Schultz, P.G. Small-molecule synergist of the Wnt/β-catenin signaling pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 7444–7448. [Google Scholar] [CrossRef]
- Takase, S.; Kurokawa, R.; Arai, D.; Kanemoto Kanto, K.; Okino, T.; Nakao, Y.; Kushiro, T.; Yoshida, M.; Matsumoto, K. A quantitative shRNA screen identifies ATP1A1 as a gene that regulates cytotoxicity by aurilide B. Sci. Rep. 2017, 7, 2002. [Google Scholar] [CrossRef]
- le Sage, C.; Lawo, S.; Panicker, P.; Scales, T.M.; Rahman, S.A.; Little, A.S.; McCarthy, N.J.; Moore, J.D.; Cross, B.C. Dual direction CRISPR transcriptional regulation screening uncovers gene networks driving drug resistance. Sci. Rep. 2017, 7, 17693. [Google Scholar] [CrossRef]
- Shendure, J.; Findlay, G.M.; Snyder, M.W. Genomic medicine–progress, pitfalls, and promise. Cell 2019, 177, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Huminiecki, L.; Horbańczuk, J.; Atanasov, A.G. The functional genomic studies of curcumin. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 107–118. [Google Scholar]
- Peng, J.; Sun, B.-F.; Chen, C.-Y.; Zhou, J.-Y.; Chen, Y.-S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.-S. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Nair, N.U.; Dinstag, G.; Chapman, L.; Chung, Y.; Wang, K.; Sinha, S.; Cha, H.; Kim, D.; Schperberg, A.V. Synthetic lethality-mediated precision oncology via the tumor transcriptome. Cell 2021, 184, 2487–2502.e13. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.; Deldalle, J.; Hallaert, C.; Robineau, O.; Ajana, F.; Choisy, P.; Hober, D.; Bocket, L. RNA and DNA Sanger sequencing versus next-generation sequencing for HIV-1 drug resistance testing in treatment-naive patients. J. Antimicrob. Chemother. 2017, 72, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dorado, G.; Gálvez, S.; Rosales, T.; Vásquez, V.; Hernández, P. Analyzing modern biomolecules: The revolution of nucleic-acid sequencing-review. Biomolecules 2021, 11, 1111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and their applications. J. Chromatogr. Sci. 2016, 55, 182–196. [Google Scholar] [CrossRef]
- Cristea, I.M.; Gaskell, S.J.; Whetton, A.D. Proteomics techniques and their application to hematology. Blood 2004, 103, 3624–3634. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Is proteomics the new genomics? Cell 2007, 130, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Haberland, V.; Baird, D.; Walker, V.; Haycock, P.C.; Hurle, M.R.; Gutteridge, A.; Erola, P.; Liu, Y.; Luo, S. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 2020, 52, 1122–1131. [Google Scholar] [CrossRef]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The emerging role of mass spectrometry-based proteomics in drug discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Marquart, J.; Chen, E.Y.; Prasad, V. Estimation of the percentage of US patients with cancer who benefit from genome-driven oncology. JAMA Oncol. 2018, 4, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Charron, G.; Zhang, M.M.; Yount, J.S.; Wilson, J.; Raghavan, A.S.; Shamir, E.; Hang, H.C. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J. Am. Chem. Soc. 2009, 131, 4967–4975. [Google Scholar] [CrossRef] [PubMed]
- Piétu, G.; Mariage-Samson, R.; Fayein, N.-A.; Matingou, C.; Eveno, E.; Houlgatte, R.; Decraene, C.; Vandenbrouck, Y.; Tahi, F.; Devignes, M.-D. The Genexpress IMAGE knowledge base of the human brain transcriptome: A prototype integrated resource for functional and computational genomics. Genome Res. 1999, 9, 195–209. [Google Scholar] [CrossRef]
- Jin, Y.; Yoon, Y.J.; Jeon, Y.J.; Choi, J.; Lee, Y.-J.; Lee, J.; Choi, S.; Nash, O.; Han, D.C.; Kwon, B.-M. Geranylnaringenin (CG902) inhibits constitutive and inducible STAT3 activation through the activation of SHP-2 tyrosine phosphatase. Biochem. Pharmacol. 2017, 142, 46–57. [Google Scholar] [CrossRef]
- Kirsch, V.C.; Orgler, C.; Braig, S.; Jeremias, I.; Auerbach, D.; Müller, R.; Vollmar, A.M.; Sieber, S.A. The cytotoxic natural product vioprolide A targets nucleolar protein 14, which is essential for ribosome biogenesis. Angew. Chem. Int. Ed. 2020, 59, 1595–1600. [Google Scholar] [CrossRef]
- Geng, J.; Liu, W.; Gao, J.; Jiang, C.; Fan, T.; Sun, Y.; Qin, Z.H.; Xu, Q.; Guo, W.; Gao, J. Andrographolide alleviates Parkinsonism in MPTP-PD mice via targeting mitochondrial fission mediated by dynamin-related protein 1. Br. J. Pharmacol. 2019, 176, 4574–4591. [Google Scholar] [CrossRef] [PubMed]
- West, G.M.; Tucker, C.L.; Xu, T.; Park, S.K.; Han, X.; Yates, J.R., III; Fitzgerald, M.C. Quantitative proteomics approach for identifying protein–drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci. USA 2010, 107, 9078–9082. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Fujii, R.; Hemmi, I.; Ueda, K.; Igeta, Y. Extracellular vesicle proteome unveils cathepsin B connection to Alzheimer’s disease pathogenesis. Brain 2024, 147, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer. Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef]
- Perrin, J.; Werner, T.; Kurzawa, N.; Rutkowska, A.; Childs, D.D.; Kalxdorf, M.; Poeckel, D.; Stonehouse, E.; Strohmer, K.; Heller, B. Identifying drug targets in tissues and whole blood with thermal-shift profiling. Nat. Biotechnol. 2020, 38, 303–308. [Google Scholar] [CrossRef]
- Lomenick, B.; Olsen, R.W.; Huang, J. Identification of direct protein targets of small molecules. ACS Chem. Biol. 2011, 6, 34–46. [Google Scholar] [CrossRef]
- Reiche, J.; Huber, O. Post-translational modifications of tight junction transmembrane proteins and their direct effect on barrier function. Biochim. Biophys. Acta BBA-Biomembr. 2020, 1862, 183330. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Metabolomics for investigating physiological and pathophysiological processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Zampieri, M.; Sekar, K.; Zamboni, N.; Sauer, U. Frontiers of high-throughput metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 15–23. [Google Scholar] [CrossRef]
- Sévin, D.C.; Sauer, U. Ubiquinone accumulation improves osmotic-stress tolerance in Escherichia coli. Nat. Chem. Biol. 2014, 10, 266–272. [Google Scholar] [CrossRef]
- Fan, S.; Gao, Y.; Zhang, H.; Huang, M.; Bi, H. Untargeted and targeted metabolomics and their applications in discovering drug targets. Prog. Pharm. Sci. 2017, 41, 263–269. [Google Scholar]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Yuan, K.; Yan, X.; Xu, L.; Liao, H.; Hao, X.; Wang, J.; Liu, H.; Chen, X.; Xie, K. LncRNA SNHG10 facilitates hepatocarcinogenesis and metastasis by modulating its homolog SCARNA13 via a positive feedback loop. Cancer Res. 2019, 79, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, H.; Zhao, Q.; Tao, C.; Qu, W.; Hou, Y.; Huang, R.; Sun, Z.; Zhu, G.; Jiang, X. Multiomics analysis reveals metabolic subtypes and identifies diacylglycerol kinase α (DGKA) as a potential therapeutic target for intrahepatic cholangiocarcinoma. Cancer Commun. 2024, 44, 226–250. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wang, H.; Li, T.; Wang, J.; Ren, Z.; Cai, N.; Ai, H.; Li, S.; Lu, Y.; Zhu, Y. Secreted MUP1 that reduced under ER stress attenuates ER stress induced insulin resistance through suppressing protein synthesis in hepatocytes. Pharmacol. Res. 2023, 187, 106585. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, Z.; Liu, J.; Song, M.; Zhang, T.; Chen, Y.; Hu, H.; Yang, P.; Li, B.; Song, X. Gefitinib and fostamatinib target EGFR and SYK to attenuate silicosis: A multi-omics study with drug exploration. Signal Transduct. Target. Ther. 2022, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Guo, F.; Wen, S.; Jin, X.; Wu, H.; Ren, D. Overexpressed integrin alpha 2 inhibits the activation of the transforming growth factor β pathway in pancreatic cancer via the TFCP2-SMAD2 axis. J. Exp. Clin. Cancer Res. 2022, 41, 73. [Google Scholar] [CrossRef]
- Reustle, A.; Di Marco, M.; Meyerhoff, C.; Nelde, A.; Walz, J.S.; Winter, S.; Kandabarau, S.; Büttner, F.; Haag, M.; Backert, L. Integrative-omics and HLA-ligandomics analysis to identify novel drug targets for ccRCC immunotherapy. Genome Med. 2020, 12, 1–24. [Google Scholar] [CrossRef]
- Liu, J.; Jing, W.; Wang, T.; Hu, Z.; Lu, H. Functional metabolomics revealed the dual-activation of cAMP-AMP axis is a novel therapeutic target of pancreatic cancer. Pharmacol. Res. 2023, 187, 106554. [Google Scholar] [CrossRef]
- Shao, X.; Ji, F.; Wang, Y.; Zhu, L.; Zhang, Z.; Du, X.; Chung, A.C.K.; Hong, Y.; Zhao, Q.; Cai, Z. Integrative chemical proteomics-metabolomics approach reveals Acaca/Acacb as direct molecular targets of PFOA. Anal. Chem. 2018, 90, 11092–11098. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Gao, Q.; Deng, Y.; Ke, Y.; Ma, E.; Yang, H.; Lin, H.; Li, H.; Yang, Y.; Gong, J. GP73 is a glucogenic hormone contributing to SARS-CoV-2-induced hyperglycemia. Nat. Metab. 2022, 4, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, J.D.; Berg, H.C.; Church, G.M. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 2004, 4, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Smith, H.O.; Adams, M.D. The sequence of the human genome. Clin. Chem. 2015, 61, 1207–1208. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Chen, J. Recent progress in mass spectrometry proteomics for biomedical research. Sci. China Life Sci. 2017, 60, 1093–1113. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, S.; Jaehnig, E.J.; Krug, K.; Kim, B.-J.; Saltzman, A.B.; Chan, D.W.; Holloway, K.R.; Anurag, M.; Huang, C.; Singh, P. Microscaled proteogenomic methods for precision oncology. Nat. Commun. 2020, 11, 532. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Krug, K.; Zhang, B.; Satpathy, S.; Clauser, K.R.; Ding, L.; Ellis, M.; Gillette, M.A.; Carr, S.A. Cancer proteogenomics: Current impact and future prospects. Nat. Rev. Cancer 2022, 22, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.-W.; Reva, B. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 2020, 182, 200–225.e35. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Roumeliotis, T.I.; Chang, Y.-H.; Chen, C.-T.; Han, C.-L.; Lin, M.-H.; Chen, H.-W.; Chang, G.-C.; Chang, Y.-L.; Wu, C.-T. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell 2020, 182, 226–244.e17. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S. Integrative proteomic characterization of human lung adenocarcinoma. Cell 2020, 182, 245–261.e17. [Google Scholar] [CrossRef]
- Ogbeide, S.; Giannese, F.; Mincarelli, L.; Macaulay, I.C. Into the multiverse: Advances in single-cell multiomic profiling. Trends Genet. 2022, 38, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, S.; Efremova, M. Method of the year 2019: Single-cell multimodal omics. Nat. Methods 2020, 17, 2020. [Google Scholar]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Baysoy, A.; Bai, Z.; Satija, R.; Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Tang, F. Recent advances in single-cell sequencing technologies. Precis. Clin. Med. 2022, 5, pbac002. [Google Scholar] [CrossRef] [PubMed]
- Nassar, S.F.; Raddassi, K.; Wu, T. Single-cell multiomics analysis for drug discovery. Metabolites 2021, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Terekhanova, N.V.; Karpova, A.; Liang, W.-W.; Strzalkowski, A.; Chen, S.; Li, Y.; Southard-Smith, A.N.; Iglesia, M.D.; Wendl, M.C.; Jayasinghe, R.G. Epigenetic regulation during cancer transitions across 11 tumour types. Nature 2023, 623, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhao, X.; Zhang, Y.; Li, Y.; Liu, S.; Han, J.; Sun, Z.; Wang, C.; Deng, D.; Wang, S. Single cell multi-omics reveal intra-cell-line heterogeneity across human cancer cell lines. Nat. Commun. 2023, 14, 8170. [Google Scholar] [CrossRef]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef]
- Eisenstein, M. Seven technologies to watch in 2022. Nature 2022, 601, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Wang, J.T.; Jayasinghe, R.G.; O’Neal, J.; Tsai, C.-F.; Rettig, M.P.; Song, Y.; Liu, R.; Zhao, Y.; Ibrahim, O.M. Single-cell discovery and multiomic characterization of therapeutic targets in multiple myeloma. Cancer Res. 2023, 83, 1214–1233. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, A.; Zhou, Y.; Chen, P.; Wang, X.; Huang, J.; Gao, J.; Wang, X.; Shu, L.; Lu, J. Spatially resolved multi-omics highlights cell-specific metabolic remodeling and interactions in gastric cancer. Nat. Commun. 2023, 14, 2692. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; William, H.G.; Xu, K.H. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef] [PubMed]
- Bressan, D.; Battistoni, G.; Hannon, G.J. The dawn of spatial omics. Science 2023, 381, eabq4964. [Google Scholar] [CrossRef] [PubMed]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-omics: Novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020, 91, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010, 38 (Suppl. 2), W695–W699. [Google Scholar] [CrossRef]
- Tateno, Y.; Imanishi, T.; Miyazaki, S.; Fukami-Kobayashi, K.; Saitou, N.; Sugawara, H.; Gojobori, T. DNA Data Bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 2002, 30, 27–30. [Google Scholar] [CrossRef]
- Ogasawara, O.; Kodama, Y.; Mashima, J.; Kosuge, T.; Fujisawa, T. DDBJ Database updates and computational infrastructure enhancement. Nucleic Acids Res. 2020, 48, D45–D50. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Griffiths-Jones, S. miRBase: The microRNA sequence database. In MicroRNA Protocols; Humana Press: Totowa, NJ, USA, 2006; pp. 129–138. [Google Scholar]
- Liu, L.; Li, Z.; Liu, C.; Zou, D.; Li, Q.; Feng, C.; Jing, W.; Luo, S.; Zhang, Z.; Ma, L. LncRNAWiki 2.0: A knowledgebase of human long non-coding RNAs with enhanced curation model and database system. Nucleic Acids Res. 2022, 50, D190–D195. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37 (Suppl. 1), D211–D215. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Lo Conte, L.; Ailey, B.; Hubbard, T.J.; Brenner, S.E.; Murzin, A.G.; Chothia, C. SCOP: A structural classification of proteins database. Nucleic Acids Res. 2000, 28, 257–259. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Hulo, N.; Bairoch, A.; Bulliard, V.; Cerutti, L.; De Castro, E.; Langendijk-Genevaux, P.S.; Pagni, M.; Sigrist, C.J. The PROSITE database. Nucleic Acids Res. 2006, 34 (Suppl. 1), D227–D230. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32 (Suppl. S1), D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35 (Suppl. 1), D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. Review The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 2015, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Tian, G.; Guo, X. TARGET database introduction and data extraction. Chin. J. Evid. Based Cardiovasc. Med. 2019, 11, 9–12. [Google Scholar]
- Zhang, J.; Bajari, R.; Andric, D.; Gerthoffert, F.; Lepsa, A.; Nahal-Bose, H.; Stein, L.D.; Ferretti, V. The international cancer genome consortium data portal. Nat. Biotechnol. 2019, 37, 367–369. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, M.; da Veiga Leprevost, F.; Squizzato, S.; Park, Y.M.; Haug, K.; Carroll, A.J.; Spalding, D.; Paschall, J.; Wang, M. Discovering and linking public omics data sets using the Omics Discovery Index. Nat. Biotechnol. 2017, 35, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Craven, K.E.; Gökmen-Polar, Y.; Badve, S.S. CIBERSORT analysis of TCGA and METABRIC identifies subgroups with better outcomes in triple negative breast cancer. Sci. Rep. 2021, 11, 4691. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddou, M.; DiStefano, M.S.; Riesel, E.A.; Carrasco, E.; Holzapfel, H.Y.; Jones, D.C.; Smith, G.R.; Stern, A.D.; Somani, S.S.; Thompson, T.V. Drug response consistency in CCLE and CGP. Nature 2016, 540, E9–E10. [Google Scholar] [CrossRef] [PubMed]
- Satterlee, J.S.; Chadwick, L.H.; Tyson, F.L.; McAllister, K.; Beaver, J.; Birnbaum, L.; Volkow, N.D.; Wilder, E.L.; Anderson, J.M.; Roy, A.L. The NIH common fund/roadmap epigenomics program: Successes of a comprehensive consortium. Sci. Adv. 2019, 5, eaaw6507. [Google Scholar] [CrossRef]
- Chadwick, L.H. The NIH roadmap epigenomics program data resource. Epigenomics 2012, 4, 317–324. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Omic Name | Classification | Common Reference Databases | Website | Main Functions | Reference |
|---|---|---|---|---|---|
| Genomics | Three major DNA Databases | GenBank | http://www.ncbi.nlm.nih.gov/genbank/ | Provides genome-wide 2D gel-electrophoresis profiles, collecting 2D gel-electrophoresis maps of proteomes of organisms with known genomic information. | [91] |
| EMBL | https://www.ebi.ac.uk | Nucleic acid sequences, genomes, microarray gene expression, protein sequences, annotations, and many other biological data | [92] | ||
| DDBJ | https://www.ddbj.nig.ac.jp/index-e.html | Genomic, transcriptomic, epigenomic, exomic, macrogenomic, macrotranscriptomic, and other multi-omics data for human, animal, and other samples. | [93,94] | ||
| Transcriptomics | NCBI | miRBase | http://www.mirbase.org | The most comprehensive miRNA database with nearly 40,000 miRNAs from more than 200 species. | [95,96] |
| EMBL-EBI | LncRNAwiki | https://ngdc.cncb.ac.cn/lncrnawiki1/index.php/Main_Page | Integration of more than 100,000 LncRNAs currently available, classification of long non-coding RNAs | [97] | |
| NGDC | Rfam | http://rfam.org | Identification of non-coding RNAs, commonly used to annotate new nucleic acid sequences or genome sequences | [98,99] | |
| Proteomics | Protein Sequence Database | UniProt | http://www.uniprot.org | Contains protein sequences, functional information, and an index of research papers. | [100] |
| PIR | https://proteininformationresource.org | Database integrating public resources on protein function prediction data | [87] | ||
| InterPro | http://www.ebi.ac.uk/interpro/ | Integrated protein structural domain and functional site databases with data resources on protein families, domains, repeat sequences, and sites of action | [101,102] | ||
| Protein Structure Database | Protein Data Bank | https://www.rcsb.org | X-ray diffraction structures, NMR spectra, electron microscopy (EM) imaging, and some special structures (e.g., DNA and RNA structure libraries) are included. | [103] | |
| SCOP | https://www.ebi.ac.uk/pdbe/scop/ | Classifies known protein structures and describes the functions and evolutionary relationships of proteins of known structure based on the amino acid composition of different proteins and similarities in tertiary structure | [104] | ||
| Proteome Database | PRIDE | https://www.ebi.ac.uk/pride/archive/ | Classifies known protein structures and describes the functions and evolutionary relationships of proteins of known structure based on the amino acid composition of different proteins, as well as similarities in tertiary structure. | [105] | |
| Protein Functional Domain Database | PROSITE | https://prosite.expasy.org | Database of protein families and structural domains containing biologically significant sites, patterns, and statistical features that can help identify protein families | [106] | |
| Protein Interaction Database | DIP | https://dip.doe-mbi.ucla.edu/dip/Main.cgi | Tool for studying biological response mechanisms | [107] | |
| Metabolomics | Metabolic Pathways Database | KEGG | https://www.kegg.jp | Linking genomic information to higher-order functional information for the study of genomes, metabolomes, signaling pathways, and biochemical reactions | [108,109] |
| GO | https://geneontology.org | Standardizing the function of genes and proteins | [110] | ||
| Metabolome commonly used Database | HMDB | https://hmdb.ca | Comprehensive reference information on human metabolites and their associated biological, physiological, and chemical properties | [111] |
| Multi-omics Database | Website | Omics Assay Type | Reference |
|---|---|---|---|
| The Cancer Genome Atlas (TCGA) | https://www.cancer.gov/ccg/research/genome-sequencing/tcga | RNA-Seq, DNA-Seq, miRNA-Seq, SNV, CNV, DNA methylation, and RPPA | [112] |
| TARGET | https://www.cancer.gov/ccg/research/genome-sequencing/target | Gene expression, miRNA expression, copy number, and sequencing data | [113] |
| International Cancer Genomics Consortium (ICGC) | https://dcc.icgc.org/ | Whole genome sequencing, genomic variation data (somatic and germline mutations) | [114] |
| OmicsDI | https://www.omicsdi.org | Proteomics, genomics, metabolomics, and transcriptomics data | [115] |
| METABRIC | https://www.cbioportal.org | Clinical features, gene expression, SNPs, and CNVs | [116] |
| CCLE | https://sites.broadinstitute.org/ccle | Gene expression, copy number, and sequencing data; pharmacological profile of 24 anticancer drugs | [117] |
| Roadmap Epigenomics | https://commonfund.nih.gov/epigenomics | RNA-seq, ChIP-seq (histones), DNase-seq, and methylation data | [118,119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, P.; Fan, R.; Zhang, N.; Wu, C.; Zhang, Y. Advances in Integrated Multi-omics Analysis for Drug-Target Identification. Biomolecules 2024, 14, 692. https://doi.org/10.3390/biom14060692
Du P, Fan R, Zhang N, Wu C, Zhang Y. Advances in Integrated Multi-omics Analysis for Drug-Target Identification. Biomolecules. 2024; 14(6):692. https://doi.org/10.3390/biom14060692
Chicago/Turabian StyleDu, Peiling, Rui Fan, Nana Zhang, Chenyuan Wu, and Yingqian Zhang. 2024. "Advances in Integrated Multi-omics Analysis for Drug-Target Identification" Biomolecules 14, no. 6: 692. https://doi.org/10.3390/biom14060692
APA StyleDu, P., Fan, R., Zhang, N., Wu, C., & Zhang, Y. (2024). Advances in Integrated Multi-omics Analysis for Drug-Target Identification. Biomolecules, 14(6), 692. https://doi.org/10.3390/biom14060692