

Roles of Mitochondrial Dysfunction in Diabetic Kidney Disease: New Perspectives from Mechanism to Therapy
Abstract
1. Introduction
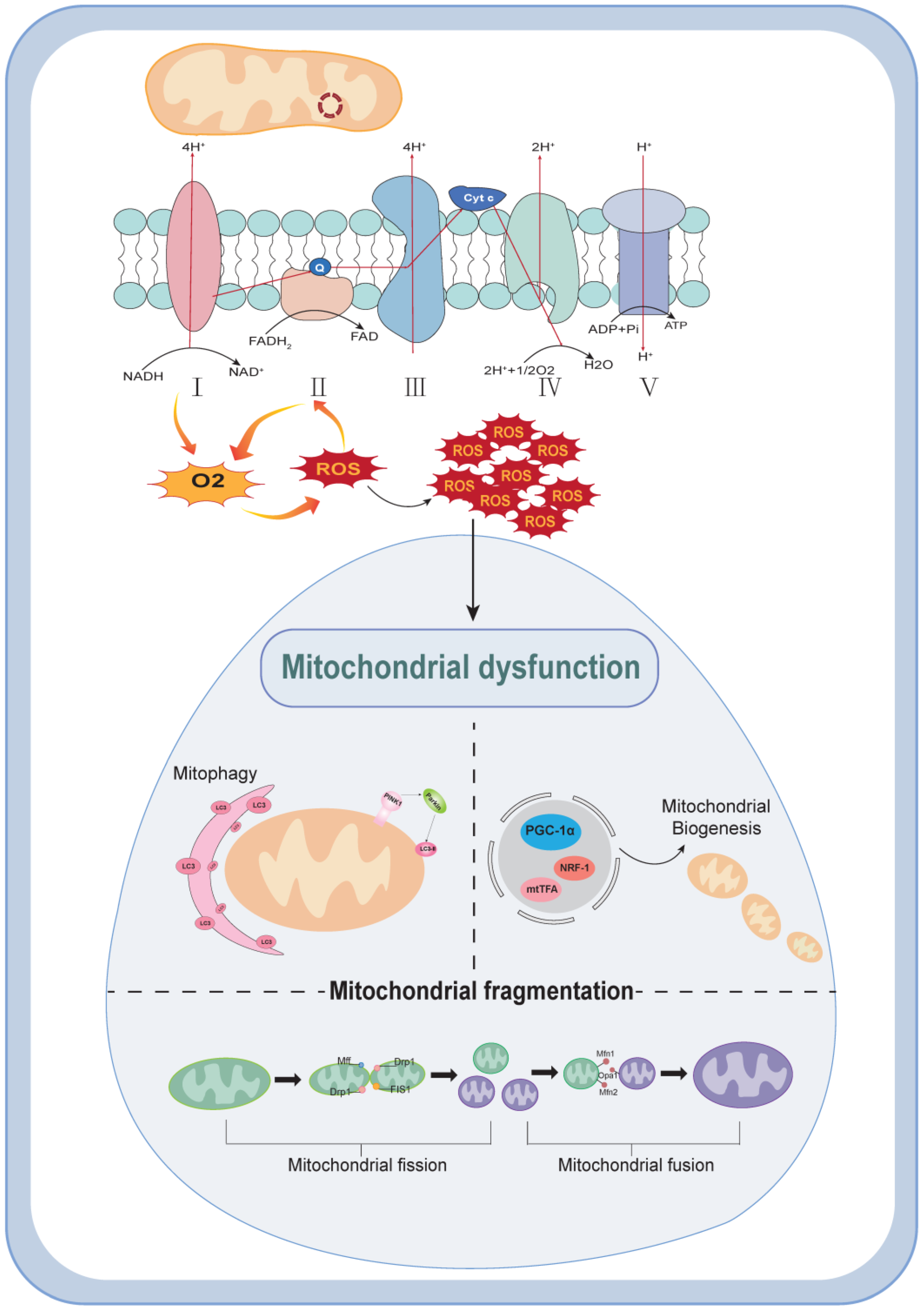
2. Mechanisms of Mitochondrial Dysfunction
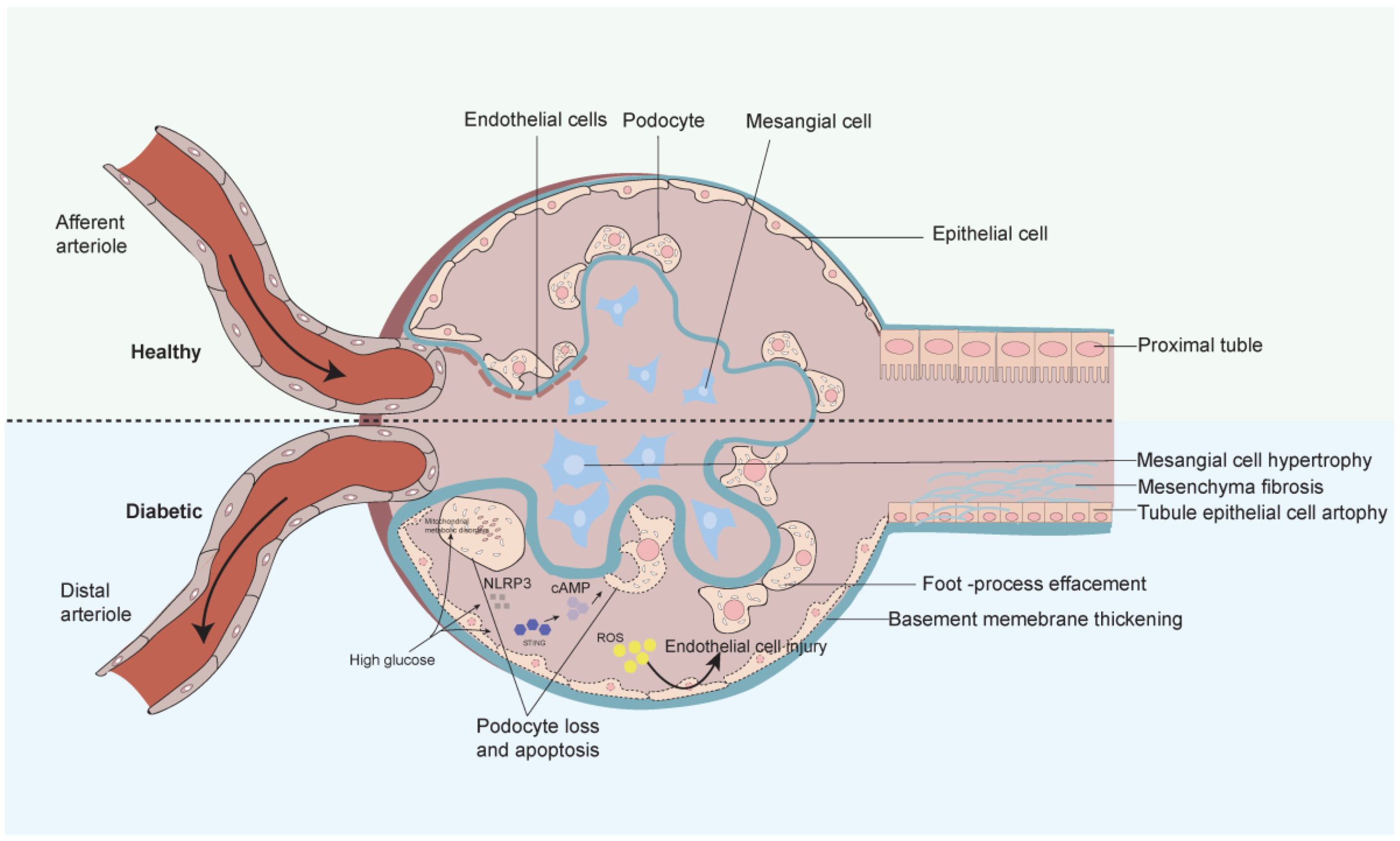
3. Progression and Mechanisms of Diabetic Nephropathy
4. Diabetic Nephropathy and Mitochondrial Dysfunction
4.1. DKD and Mitochondrial Biogenesis
4.2. DKD and ROS Generation
4.3. DKD and Mitochondrial Autophagy Disorders
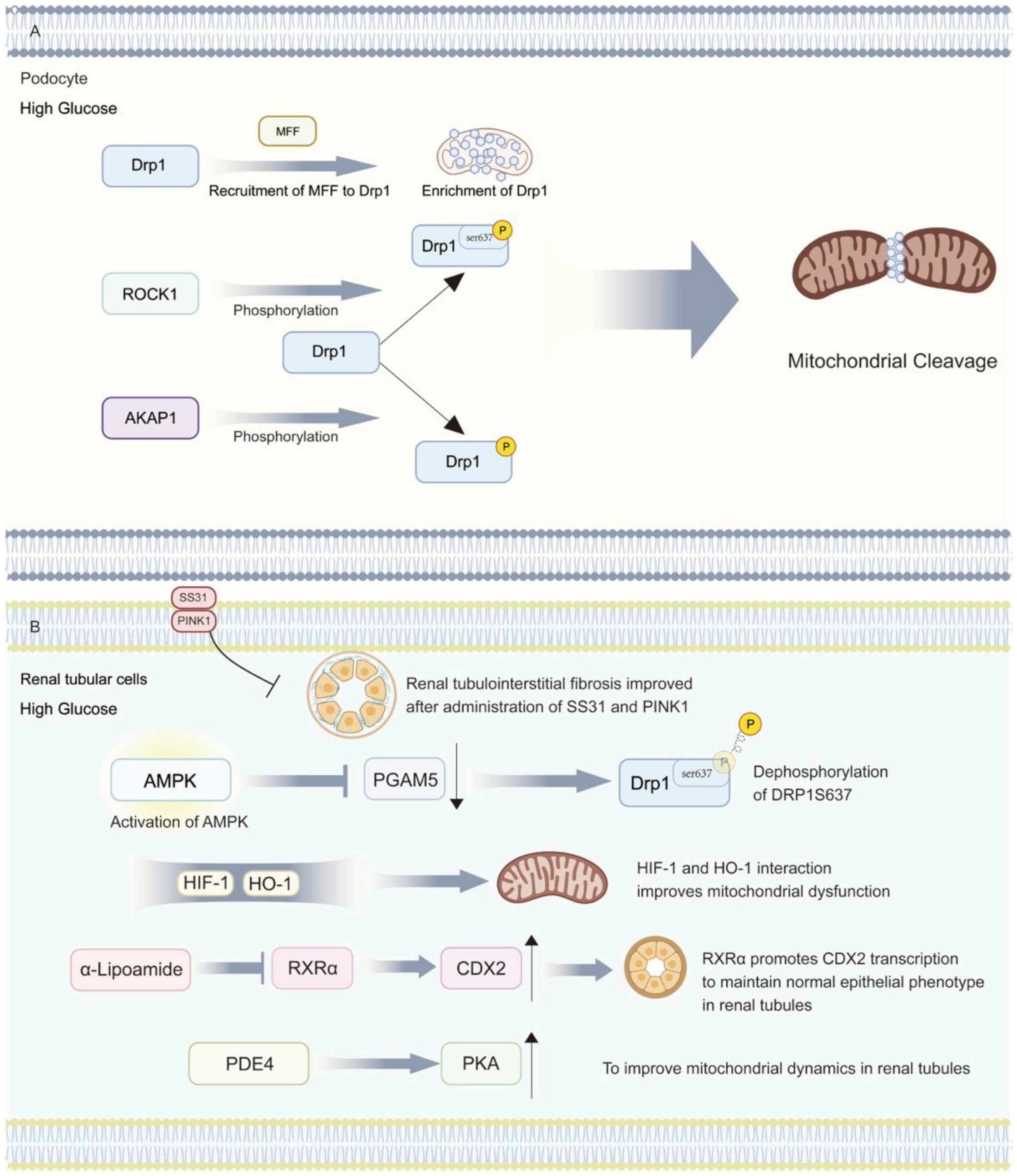
4.4. DKD and Mitochondrial Dynamics Disorders
5. Therapeutic Options Targeting Mitochondrial Dysfunction in Diabetic Nephropathy
6. Conclusions and Expectations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pan, C.Y.; Ho, L.T.; Soegondo, S.; Prodjosudjadi, W.; Suwanwalaikorn, S.; Lim, S.C.; Chan, T.M.; Chow, K.W.; Thoenes, M.; Choi, D.S. Prevalence of albuminuria and cardiovascular risk profile in a referred cohort of patients with type 2 diabetes: An Asian perspective. Diabetes Technol. Ther. 2008, 10, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.; Edi, R. Diabetic Kidney Disease: Diagnosis, Treatment, and Prevention. Am. Fam. Physician 2019, 99, 751–759. [Google Scholar] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 353–389. [Google Scholar] [CrossRef]
- Zhang, H.F.; Liu, H.M.; Xiang, J.Y.; Zhou, X.C.; Wang, D.; Chen, R.Y.; Tan, W.L.; Liang, L.Q.; Liu, L.L.; Shi, M.J.; et al. Alpha lipoamide inhibits diabetic kidney fibrosis via improving mitochondrial function and regulating RXRα expression and activation. Acta Pharmacol. Sin. 2023, 44, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Y.; Liu, Z.; He, L. Adiponectin promotes repair of renal tubular epithelial cells by regulating mitochondrial biogenesis and function. Metabolism 2022, 128, 154959. [Google Scholar] [CrossRef]
- Tao, L.C.; Wang, T.T.; Zheng, L.; Hua, F.; Li, J.J. The Role of Mitochondrial Biogenesis Dysfunction in Diabetic Cardiomyopathy. Biomol. Ther. 2022, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Siragy, H.M. Pro-renin receptor suppresses mitochondrial biogenesis and function via AMPK/SIRT-1/ PGC-1α pathway in diabetic kidney. PLoS ONE 2019, 14, e0225728. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884s–890s. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, B.; Liao, Z.; Xue, M.; Zou, Z.; Feng, J.; Sheng, J. DUSP1 overexpression attenuates renal tubular mitochondrial dysfunction by restoring Parkin-mediated mitophagy in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2021, 559, e141–147. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett. 2010, 584, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J. Cell. Physiol. 2019, 234, 3043–3057. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; López Del Amo, V.; Arndt, S.; Bueno, D.; Tenzer, S.; Hanschmann, E.M.; Berndt, C.; Methner, A. Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery. Cells 2020, 9, 815. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Liu, Q.; Liu, B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Ma, Y.; Chen, Z.; Hu, J.; Yang, Q.; Ding, G. Mitochondrial pyruvate carrier 2 mediates mitochondrial dysfunction and apoptosis in high glucose-treated podocytes. Life Sci. 2019, 237, 116941. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Gong, X.; Liu, X.; Zhang, R.; Wang, Y.; Huang, B.; Zhang, L.; Zheng, H.; Zheng, Y. Deficiency of Mitochondrial Glycerol 3-Phosphate Dehydrogenase Exacerbates Podocyte Injury and the Progression of Diabetic Kidney Disease. Diabetes 2021, 70, 1372–1387. [Google Scholar] [CrossRef] [PubMed]
- Zang, N.; Cui, C.; Guo, X.; Song, J.; Hu, H.; Yang, M.; Xu, M.; Wang, L.; Hou, X.; He, Q.; et al. cGAS-STING activation contributes to podocyte injury in diabetic kidney disease. iScience 2022, 25, 105145. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, Y.; Xie, D.; Jiang, H. ManNAc protects against podocyte pyroptosis via inhibiting mitochondrial damage and ROS/NLRP3 signaling pathway in diabetic kidney injury model. Int. Immunopharmacol. 2022, 107, 108711. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Khramova, A.; Boi, R.; Fridén, V.; Granqvist, A.B.; Nilsson, U.; Tenstad, O.; Oveland, E.; Haraldsson, B.; Ebefors, K.; Nyström, J. Proteoglycans contribute to the functional integrity of the glomerular endothelial cell surface layer and are regulated in diabetic kidney disease. Sci. Rep. 2021, 11, 8487. [Google Scholar] [CrossRef]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Renal. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef]
- Nauta, F.L.; Boertien, W.E.; Bakker, S.J.; van Goor, H.; van Oeveren, W.; de Jong, P.E.; Bilo, H.; Gansevoort, R.T. Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care 2011, 34, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Mohandes, S.; Doke, T.; Hu, H.; Mukhi, D.; Dhillon, P.; Susztak, K. Molecular pathways that drive diabetic kidney disease. J. Clin. Investig. 2023, 133, e165654. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, Z.; Li, H.; Fan, J.; Yang, S.; Chen, C.; Wang, D.W. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell 2017, 16, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ajay, A.K.; Chang, J.H.; Mou, S.; Zhao, H.; Kishi, S.; Li, J.; Brooks, C.R.; Xiao, S.; Woo, H.M.; et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021, 33, 1042–1061.e1047. [Google Scholar] [CrossRef] [PubMed]
- Salti, T.; Khazim, K.; Haddad, R.; Campisi-Pinto, S.; Bar-Sela, G.; Cohen, I. Glucose Induces IL-1α-Dependent Inflammation and Extracellular Matrix Proteins Expression and Deposition in Renal Tubular Epithelial Cells in Diabetic Kidney Disease. Front. Immunol. 2020, 11, 1270. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Ma, Y.; Chen, Z.; Hu, J.; Chen, C.; Fan, Y.; Liang, W.; Ding, G. Sestrin-2 regulates podocyte mitochondrial dysfunction and apoptosis under high-glucose conditions via AMPK. Int. J. Mol. Med. 2020, 45, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, M.; Liu, Y.; Pang, Y.; Tian, L.; Zhao, J.; Liu, M.; Shen, C.; Meng, Y.; Wang, Y.; et al. BaoShenTongLuo formula protects against podocyte injury by regulating AMPK-mediated mitochondrial biogenesis in diabetic kidney disease. Chin. Med. 2023, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef]
- Shen, Q.; Fang, J.; Guo, H.; Su, X.; Zhu, B.; Yao, X.; Wang, Y.; Cao, A.; Wang, H.; Wang, L. Astragaloside IV attenuates podocyte apoptosis through ameliorating mitochondrial dysfunction by up-regulated Nrf2-ARE/TFAM signaling in diabetic kidney disease. Free Radic. Biol. Med. 2023, 203, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Sestrin2 Signaling Pathway Regulates Podocyte Biology and Protects against Diabetic Nephropathy. J. Diabetes Res. 2023, 2023, 8776878. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Yang, X.; Gu, X.; Chu, L.; Sun, S.; Sun, J.; Song, P.; Mu, Q.; Wang, Y.; Sun, X.; et al. CUL3 induces mitochondrial dysfunction via MRPL12 ubiquitination in renal tubular epithelial cells. Febs J. 2023, 290, 5340–5352. [Google Scholar] [CrossRef] [PubMed]
- Ala, M.; Khoshdel, M.R.F.; Dehpour, A.R. Empagliflozin Enhances Autophagy, Mitochondrial Biogenesis, and Antioxidant Defense and Ameliorates Renal Ischemia/Reperfusion in Nondiabetic Rats. Oxid. Med. Cell. Longev. 2022, 2022, 1197061. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhou, Q.; Liu, C.; Zeng, Y.; Yuan, S. Klotho deficiency aggravates diabetes-induced podocyte injury due to DNA damage caused by mitochondrial dysfunction. Int. J. Med. Sci. 2020, 17, 2763–2772. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chen, Z.; Ma, Y.; Yang, X.; Zhu, Z.; Zhang, Z.; Hu, J.; Liang, W.; Ding, G. AKAP1 contributes to impaired mtDNA replication and mitochondrial dysfunction in podocytes of diabetic kidney disease. Int. J. Biol. Sci. 2022, 18, 4026–4042. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jiang, M.; Zhao, Y.; Gong, J.; Su, H.; Yuan, F.; Fang, K.; Yuan, X.; Yu, X.; Dong, H.; et al. Berberine protects against diabetic kidney disease via promoting PGC-1α-regulated mitochondrial energy homeostasis. Br. J. Pharmacol. 2020, 177, 3646–3661. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Yang, Z.; Zhang, C.; Shi, Y.; Han, W.; Song, S.; Mu, L.; Du, C.; Shi, Y. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism 2021, 118, 154748. [Google Scholar] [CrossRef] [PubMed]
- He, J.Y.; Hong, Q.; Chen, B.X.; Cui, S.Y.; Liu, R.; Cai, G.Y.; Guo, J.; Chen, X.M. Ginsenoside Rb1 alleviates diabetic kidney podocyte injury by inhibiting aldose reductase activity. Acta Pharmacol. Sin. 2022, 43, 342–353. [Google Scholar] [CrossRef]
- Woo, C.Y.; Baek, J.Y.; Kim, A.R.; Hong, C.H.; Yoon, J.E.; Kim, H.S.; Yoo, H.J.; Park, T.S.; Kc, R.; Lee, K.U.; et al. Inhibition of Ceramide Accumulation in Podocytes by Myriocin Prevents Diabetic Nephropathy. Diabetes Metab. J. 2020, 44, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Q.; Kong, D.; Long, Z.; Guo, Y.; Wang, S.; Liu, R.; Hai, C. Down-regulation of SETD6 protects podocyte against high glucose and palmitic acid-induced apoptosis, and mitochondrial dysfunction via activating Nrf2-Keap1 signaling pathway in diabetic nephropathy. J. Mol. Histol. 2020, 51, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Tsogbadrakh, B.; Yang, S.; Ryu, H.; Kang, E.; Kang, M.; Kang, H.G.; Ahn, C.; Oh, K.H. Klotho ameliorates diabetic nephropathy via LKB1-AMPK-PGC1α-mediated renal mitochondrial protection. Biochem. Biophys. Res. Commun. 2021, 534, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Liu, Y.; Wang, N.; Zhen, J.; Zhang, B.; Hou, S.; Cui, Z.; Wan, Q.; Feng, H. Transcription of MRPL12 regulated by Nrf2 contributes to the mitochondrial dysfunction in diabetic kidney disease. Free Radic. Biol. Med. 2021, 164, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Kim, J.Y.; Ahn, E.; Hyeon, J.S.; Kim, G.H.; Park, K.J.; Jung, Y.; Lee, Y.J.; Son, M.K.; Kim, S.W.; et al. Associations between local acidosis induced by renal LDHA and renal fibrosis and mitochondrial abnormalities in patients with diabetic kidney disease. Transl. Res. 2022, 249, 88–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L.; Zhao, J.; Guo, X.; Luo, Y.; Hu, W.; Zhao, T. Tumor Necrosis Factor Receptor-Associated Protein 1 Protects against Mitochondrial Injury by Preventing High Glucose-Induced mPTP Opening in Diabetes. Oxid. Med. Cell. Longev. 2020, 2020, 6431517. [Google Scholar] [CrossRef]
- Yao, L.; Liang, X.; Liu, Y.; Li, B.; Hong, M.; Wang, X.; Chen, B.; Liu, Z.; Wang, P. Non-steroidal mineralocorticoid receptor antagonist finerenone ameliorates mitochondrial dysfunction via PI3K/Akt/eNOS signaling pathway in diabetic tubulopathy. Redox Biol. 2023, 68, 102946. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.; Manoharan, J.; Gupta, A.; Gupta, D.; Elwakiel, A.; Khawaja, H.; Fatima, S.; Zimmermann, S.; Singh, K.; Ambreen, S.; et al. Activated Protein C Ameliorates Tubular Mitochondrial Reactive Oxygen Species and Inflammation in Diabetic Kidney Disease. Nutrients 2022, 14, 3138. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Gan, Y.; Huang, W.F.; Wu, H.L.; Zhang, X.Q.; Zheng, H.J.; Liu, H.F. Lysosome restoration to activate podocyte autophagy: A new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019, 10, 806. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Fan, Y.; Feng, J.; Zhu, Z.; Luo, Z.; Hu, H.; Li, W.; Yang, H.; Ding, G. ALCAT1-mediated abnormal cardiolipin remodelling promotes mitochondrial injury in podocytes in diabetic kidney disease. Cell Commun. Signal. 2024, 22, 26. [Google Scholar] [CrossRef]
- Woo, C.Y.; Kc, R.; Kim, M.; Kim, H.S.; Baek, J.Y.; Koh, E.H. Autophagic flux defect in diabetic kidney disease results in megamitochondria formation in podocytes. Biochem. Biophys. Res. Commun. 2020, 521, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Yan, W.; Guo, T.; Liu, N.; Wang, Z.; Shang, J.; Wei, X.; Cui, X.; Sun, Y.; Ren, S.; et al. Erythropoietin Mitigates Diabetic Nephropathy by Restoring PINK1/Parkin-Mediated Mitophagy. Front. Pharmacol. 2022, 13, 883057. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.S.; Chen, X.M.; Hua, W.; He, J.L.; Liu, T.; Li, X.J.; Wan, J.M.; Gan, H.; Du, X.G. PINK1/Parkin mediated mitophagy ameliorates palmitic acid-induced apoptosis through reducing mitochondrial ROS production in podocytes. Biochem. Biophys. Res. Commun. 2020, 525, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Jiang, L.; Li, Y.Y.; Huang, Y.B.; Hu, X.R.; Zhu, W.; Wang, X.; Wu, Y.G.; Meng, X.M.; Qi, X.M. Wogonin protects glomerular podocytes by targeting Bcl-2-mediated autophagy and apoptosis in diabetic kidney disease. Acta Pharmacol. Sin. 2022, 43, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.; Luo, D.; Li, Y.; Li, Y.; Wang, Q.; Hu, Z.; Ye, Z.; Peng, H. Irisin ameliorates diabetic kidney disease by restoring autophagy in podocytes. FASEB J. 2023, 37, e23175. [Google Scholar] [CrossRef] [PubMed]
- Salemkour, Y.; Yildiz, D.; Dionet, L.; t Hart, D.C.; Verheijden, K.A.T.; Saito, R.; Mahtal, N.; Delbet, J.D.; Letavernier, E.; Rabant, M.; et al. Podocyte Injury in Diabetic Kidney Disease in Mouse Models Involves TRPC6-mediated Calpain Activation Impairing Autophagy. J. Am. Soc. Nephrol. 2023, 34, 1823–1842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Luan, G.; Peng, S.; Fang, Y.; Fang, Q.; Shen, S.; Wu, K.; Qian, S.; Jia, W.; Ye, J.; et al. Huangkui capsule attenuates diabetic kidney disease through the induction of mitophagy mediated by STING1/PINK1 signaling in tubular cells. Phytomedicine 2023, 119, 154975. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bai, F.; Song, H.; Xiao, R.; Wang, Y.; Yang, H.; Ren, X.; Li, S.; Gao, L.; Ma, C.; et al. Upregulation of TIPE1 in tubular epithelial cell aggravates diabetic nephropathy by disrupting PHB2 mediated mitophagy. Redox Biol. 2022, 50, 102260. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.S.; Xiang, X.Y.; Chen, X.M.; He, J.L.; Liu, T.; Gan, H.; Du, X.G. Inhibition of soluble epoxide hydrolase attenuates renal tubular mitochondrial dysfunction and ER stress by restoring autophagic flux in diabetic nephropathy. Cell Death Dis. 2020, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Liang, T.; Lin, J.; Xie, J.; Cao, D.; Wang, H.; Li, Q.; Li, S.; Li, J.; Zhang, Y.; et al. Linc279227 contributes to mitochondrial dysfunction in high glucose-treated mouse renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2023, 644, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Kong, Z.L.; Che, K.; Hu, J.X.; Li, Y.; Huang, Y.J.; Guo, H.; Qi, M.M.; Chi, J.W.; Wang, Y.G. The role of mitochondrial fission factor in podocyte injury in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2022, 624, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ma, Y.; Yang, Q.; Hu, J.; Feng, J.; Liang, W.; Ding, G. AKAP1 mediates high glucose-induced mitochondrial fission through the phosphorylation of Drp1 in podocytes. J. Cell. Physiol. 2020, 235, 7433–7448. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, X.; Wen, R.; Chen, L.; Yang, Q.; Song, S.; Xiao, G.; Su, Z.; Wang, C. Increased thromboxane/prostaglandin receptors contribute to high glucose-induced podocyte injury and mitochondrial fission through ROCK1-Drp1 signaling. Int. J. Biochem. Cell Biol. 2022, 151, 106281. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Guo, Y.; Dong, H.; Wu, F.; He, Q.; Gong, J.; Lu, F. Modified Hu-lu-ba-wan protects diabetic glomerular podocytes via promoting PKM2-mediated mitochondrial dynamic homeostasis. Phytomedicine 2024, 123, 155247. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xie, W.; Wang, X.; Luo, J.; Zhou, Y.; Cao, H.; Sun, Q.; Jiang, L.; Yang, J. SS31 Ameliorates Podocyte Injury via Inhibiting OMA1-Mediated Hydrolysis of OPA1 in Diabetic Kidney Disease. Front. Pharmacol. 2021, 12, 707006. [Google Scholar] [CrossRef]
- Tagaya, M.; Kume, S.; Yasuda-Yamahara, M.; Kuwagata, S.; Yamahara, K.; Takeda, N.; Tanaka, Y.; Chin-Kanasaki, M.; Nakae, Y.; Yokoi, H.; et al. Inhibition of mitochondrial fission protects podocytes from albumin-induced cell damage in diabetic kidney disease. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166368. [Google Scholar] [CrossRef]
- Yang, S.K.; Li, A.M.; Han, Y.C.; Peng, C.H.; Song, N.; Yang, M.; Zhan, M.; Zeng, L.F.; Song, P.A.; Zhang, W.; et al. Mitochondria-Targeted Peptide SS31 Attenuates Renal Tubulointerstitial Injury via Inhibiting Mitochondrial Fission in Diabetic Mice. Oxid. Med. Cell. Longev. 2019, 2019, 2346580. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.J.; An, H.J.; Ha, M.H.; Park, S.H.; Jeong, H.Y.; Baek, J.; Lee, S.H.; Lee, Y.H.; Lee, S.Y. PTEN-induced kinase 1 exerts protective effects in diabetic kidney disease by attenuating mitochondrial dysfunction and necroptosis. Int. J. Biol. Sci. 2023, 19, 5145–5159. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, C.; Xu, L.; Li, X.; Sun, H.; Xue, M.; Li, T.; Yu, X.; Sun, B.; Chen, L. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism 2020, 111, 154334. [Google Scholar] [CrossRef]
- Jiang, N.; Zhao, H.; Han, Y.; Li, L.; Xiong, S.; Zeng, L.; Xiao, Y.; Wei, L.; Xiong, X.; Gao, P.; et al. HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif. 2020, 53, e12909. [Google Scholar] [CrossRef]
- Zhu, X.; Deng, Z.; Cao, Y.; Zhou, Z.; Sun, W.; Liu, C.; Fan, S.; Yin, X.X. Resveratrol prevents Drp1-mediated mitochondrial fission in the diabetic kidney through the PDE4D/PKA pathway. Phytother. Res. 2023, 37, 5916–5931. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef] [PubMed]
- Barutta, F.; Bellini, S.; Gruden, G. Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin. Sci. 2022, 136, 493–520. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yang, Q.; Yang, Y.; Gao, Z.; Ma, Y.; Zhang, L.; Liang, W.; Ding, G. Sirt6 Suppresses High Glucose-Induced Mitochondrial Dysfunction and Apoptosis in Podocytes through AMPK Activation. Int. J. Biol. Sci. 2019, 15, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Liang, X.; Qiao, Y.; Chen, B.; Wang, P.; Liu, Z. Mitochondrial dysfunction in diabetic tubulopathy. Metabolism 2022, 131, 155195. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, F.; Guo, D.; Du, C.; Zhao, S.; Li, J.; Yan, Z.; Hao, J. Schisandrin B Alleviates Renal Tubular Cell Epithelial-Mesenchymal Transition and Mitochondrial Dysfunction by Kielin/Chordin-like Protein Upregulation via Akt Pathway Inactivation and Adenosine 5′-Monophosphate (AMP)-Activated Protein Kinase Pathway Activation in Diabetic Kidney Disease. Molecules 2023, 28, 7851. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Anton, M.I.; Floria, M.; Seritean Isac, P.N.; Hurjui, L.L.; Tarniceriu, C.C.; Costea, C.F.; Ciocoiu, M.; Rezus, C. Oxidative Stress and NRF2/KEAP1/ARE Pathway in Diabetic Kidney Disease (DKD): New Perspectives. Biomolecules 2022, 12, 1227. [Google Scholar] [CrossRef] [PubMed]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Myakala, K.; Wang, X.X.; Shults, N.V.; Krawczyk, E.; Jones, B.A.; Yang, X.; Rosenberg, A.Z.; Ginley, B.; Sarder, P.; Brodsky, L.; et al. NAD metabolism modulates inflammation and mitochondria function in diabetic kidney disease. J. Biol. Chem. 2023, 299, 104975. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Li, K.; Peng, X.; Kan, Y.; Li, H.; Zhu, Y.; Wang, Z.; Li, Z.; Liu, H.Y.; Cai, D. Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients 2023, 15, 4772. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Zhang, H.; Zhou, X.; Wang, D.; Chen, R.; Tan, W.; Liang, L.; Shi, M.; Zhang, F.; Xiao, Y.; et al. Atorvastatin Restores PPARα Inhibition of Lipid Metabolism Disorders by Downregulating miR-21 Expression to Improve Mitochondrial Function and Alleviate Diabetic Nephropathy Progression. Front. Pharmacol. 2022, 13, 819787. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Takashi, Y.; Muta, Y.; Oda, N.; Nagata, D.; Takahashi, H.; Tanabe, M. Mineralocorticoid Receptor Antagonists in Diabetic Kidney Disease. Front. Pharmacol. 2021, 12, 754239. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.; Palygin, O.; El-Meanawy, A.; Mattson, D.L.; Geurts, A.M.; Staruschenko, A.; Sorokin, A. p66Shc-mediated hydrogen peroxide production impairs nephrogenesis causing reduction of number of glomeruli. Life Sci. 2021, 279, 119661. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yu, H.; Sun, Q.; Pei, F.; Xia, Q.; Gao, Z.; Li, X. Grape seed proanthocyanidin extract targets p66Shc to regulate mitochondrial biogenesis and dynamics in diabetic kidney disease. Front. Pharmacol. 2022, 13, 1035755. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, T.; Zheng, X.; Cui, W.; Shang, J.; Zhao, Z. Gut microbiota, key to unlocking the door of diabetic kidney disease. Nephrology 2021, 26, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Rizk, F.H.; El Saadany, A.A.; Atef, M.M.; Abd-Ellatif, R.N.; El-Guindy, D.M.; Abdel Ghafar, M.T.; Shalaby, M.M.; Hafez, Y.M.; Mashal, S.S.A.; Basha, E.H.; et al. Ulinastatin ameliorated streptozotocin-induced diabetic nephropathy: Potential effects via modulating the components of gut-kidney axis and restoring mitochondrial homeostasis. Pflugers Arch. 2023, 475, 1161–1176. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Fang, T.; Sun, H.; Cheng, Y.; Li, T.; Xu, C.; Tang, C.; Liu, X.; Sun, B.; Chen, L. PACS-2 attenuates diabetic kidney disease via the enhancement of mitochondria-associated endoplasmic reticulum membrane formation. Cell Death Dis. 2021, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Kidney Cells | Abnormalities Caused by Mitochondrial Dysfunction | References |
|---|---|---|
| Mesenchymal cells | Metabolic disorders Increased oxidative stress Fibrosis | [80] |
| Glomerular endothelial cells | Inflammatory response Endothelial barrier damage | [81,82] |
| Podocytes | Impaired filtration function Increased apoptosis Cytoskeletal changes | [47,83,84] |
| Renal tubular cells | Impaired reabsorption Cell damage and apoptosis Metabolic imbalances | [85] |
| Mechanism | DKD Model | ||
|---|---|---|---|
| Mitochondrial biogenesis and dynamics | BaoShenTongLuo (BSTL) | BTSL restored phosphorylation of AMPK and reduced podocyte apoptosis, suppressed excessive cellular ROS production, and reversed the decrease in MMP that was observed under HG conditions | db/db mice and mouse podocytes line MPC-5 |
| Schisandrin B (Sch B) | Reduction of tubular fibrosis progression by binding to the AMPKSer172 phosphorylation site | db/db mice and the human proximal tubular cell line HK-2 | |
| Grape seed proanthocyanidin extract | Target and downregulate p66Shc | Streptozotocin-induced diabetic mice | |
| Mitochondria-targeted antioxidant | Astragaloside IV | Ameliorate mitochondrial dysfunction by up-regulated Nrf2-ARE/TFAM signaling | Mouse podocytes line |
| MitoQ | Regulation of the Nrf2/PINK1 signaling pathway | db/db mice and the human proximal tubular cell line HK-2 | |
| Inhibitors of mitochondrial inflammation | Nicotinamide riboside (NR) | Reduced cGAS-STING activation in DKD | db/db mice |
| Atorvastatin | Downregulation of miR-21 expression activates PPAR-α to enhance FAO to ameliorate tubular fibrosis | Streptozotocin-induced diabetic mice and mouse renal tubular epithelial cells | |
| Promotes mitochondrial autophagy | finerenone(FIN) | Improvement of mitochondrial autophagy via PI3K/Akt/eNOS signaling pathway | Streptozotocin-induced diabetic mice and the human proximal tubular cell line HK-2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Liu, J.; Shi, Q.; Guo, B.; Jia, H.; Yang, Y.; Fu, S. Roles of Mitochondrial Dysfunction in Diabetic Kidney Disease: New Perspectives from Mechanism to Therapy. Biomolecules 2024, 14, 733. https://doi.org/10.3390/biom14060733
Yang Y, Liu J, Shi Q, Guo B, Jia H, Yang Y, Fu S. Roles of Mitochondrial Dysfunction in Diabetic Kidney Disease: New Perspectives from Mechanism to Therapy. Biomolecules. 2024; 14(6):733. https://doi.org/10.3390/biom14060733
Chicago/Turabian StyleYang, Yichen, Jiahui Liu, Qiling Shi, Buyu Guo, Hanbing Jia, Yuxuan Yang, and Songbo Fu. 2024. "Roles of Mitochondrial Dysfunction in Diabetic Kidney Disease: New Perspectives from Mechanism to Therapy" Biomolecules 14, no. 6: 733. https://doi.org/10.3390/biom14060733
APA StyleYang, Y., Liu, J., Shi, Q., Guo, B., Jia, H., Yang, Y., & Fu, S. (2024). Roles of Mitochondrial Dysfunction in Diabetic Kidney Disease: New Perspectives from Mechanism to Therapy. Biomolecules, 14(6), 733. https://doi.org/10.3390/biom14060733