
Bile Acids as Emerging Players at the Intersection of Steatotic Liver Disease and Cardiovascular Diseases
 ,
,
Abstract
:1. Introduction
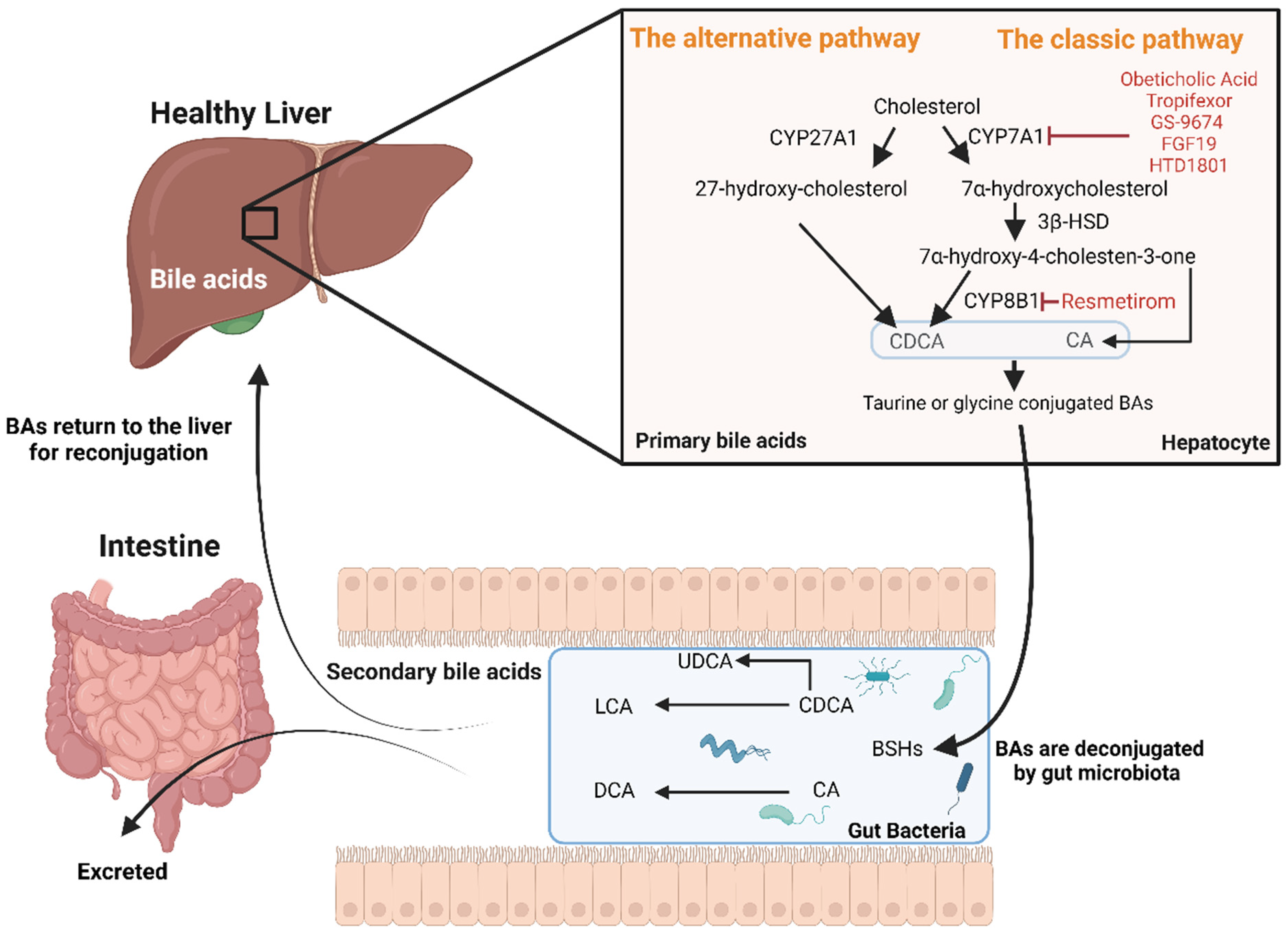
2. Bile Acid Synthesis, Regulation, and Key Signalling Pathways
- The “classic” natural pathway (quantitatively more important) in which, firstly, cholesterol is converted by the enzyme cholesterol 7α-hydroxylase (CYP7A1) to 7α-hydroxycholesterol. Secondly, the enzyme 3β-hydroxy-Δ5-C27-steroid dehydrogenase/isomerase reduces 7α-hydroxycholesterol to 7α-hydroxy-4-cholesten-3-one. Thirdly, 7α-hydroxy-4-cholesten-3-one forms chenodeoxycholic acid (CDCA) or/and cholic acid (CA) through the action of sterol 12α-hydroxylase (CYP8B1).
- The “alternative” acidic pathway in which cholesterol is converted to 27-hydroxy-cholesterol by CYP27A1 and proceeds via 3β,7α-dihydroxy-5-cholestenoic acid to form CDCA [10].
- Increased intestinal permeability with decreased expression of tight junctions. This allows for the translocation of endotoxin products (i.e., LPS from Gram-negative bacterial membrane) from the intestine to the bloodstream and thus to the portal vein and into the liver, causing inflammation and cytokine production [14].
- Apoptosis of enterocytes and hepatocytes. The hydrophobicity of secondary bile acids allows for their interaction with the phospholipids in the cell membranes of hepatocytes and enterocytes. Secondary bile acids can also travel to the cytosol inducing perturbations of the mitochondrial membrane. The alterations of the mitochondria stimulate the production of reactive oxygen species that, in turn, enhance mitochondrial permeability and cause the release of cytochrome c and other factors that form the apoptosome, ultimately causing cellular apoptosis and necrosis [15].
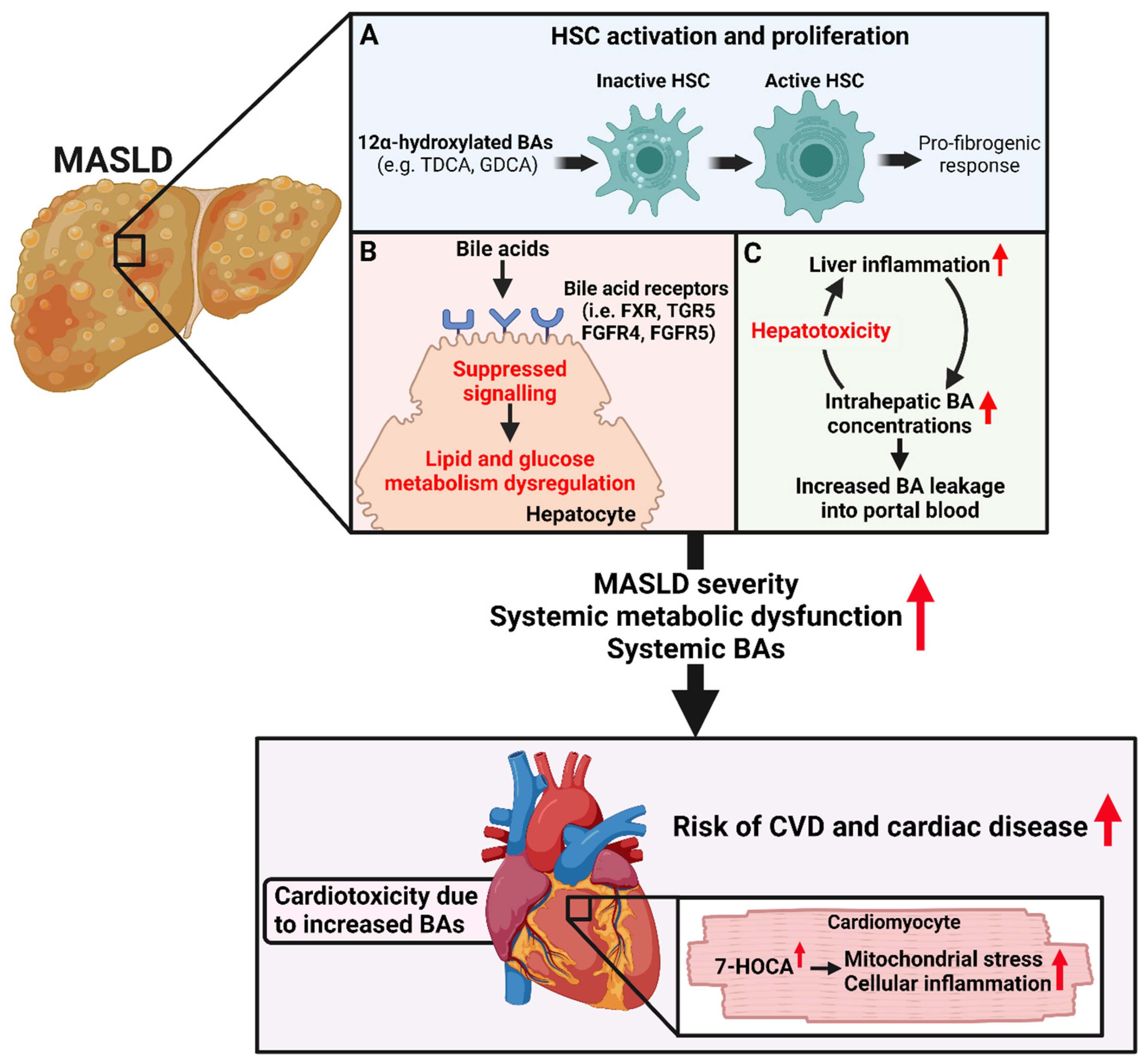
3. The Role of Trans-Genomic Bile Acid Metabolism in the Pathogenesis and Progression of MASLD
3.1. The Associations between Bile Acid Concentrations and MASLD Severity
3.2. The Role of the Gut-Microbiota-Bile Acid Axis in MASLD Pathophysiology
4. Association between Bile Acids and Cardiovascular and Cardiac Diseases
5. The Potential Role of BAs in Linking SLD and CVD and Cardiac Disease
6. Therapeutic Potential of BAs and BA Receptor Modulators to Tackle SLD and CVD
7. Future Perspectives and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.; Jamialahmadi, O.; Pelusi, S.; Baselli, G.; Dongiovanni, P.; Zanoni, I.; Santoro, L.; Maier, S.; Liguori, A.; Meroni, M.; et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021, 74, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.Q.; Jin, Y.; Wang, T.Y.; Zheng, K.I.; Rios, R.S.; Zhang, H.Y.; Targher, G.; Byrne, C.D.; Yuan, W.J.; Zheng, M.H. MAFLD and risk of CKD. Metabolism 2021, 115, 154433. [Google Scholar] [CrossRef]
- Dinani, A.; Sanyal, A. Nonalcoholic fatty liver disease: Implications for cardiovascular risk. Cardiovasc. Endocrinol. 2017, 6, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Jin, L.; Huang, W. Bile Acids, Intestinal Barrier Dysfunction, and Related Diseases. Cells 2023, 12, 1888. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Haas, J.; Grzych, G.; Tailleux, A.; Staels, B. Bile acid alterations in nonalcoholic fatty liver disease, obesity, insulin resistance and type 2 diabetes: What do the human studies tell? Curr. Opin. Lipidol. 2019, 30, 244–254. [Google Scholar] [CrossRef]
- Clayton, P.T. Disorders of bile acid synthesis. J. Inherit. Metab. Dis. 2011, 34, 593–604. [Google Scholar] [CrossRef]
- Larabi, A.B.; Masson, H.L.P.; Bäumler, A.J. Bile acids as modulators of gut microbiota composition and function. Gut Microbes 2023, 15, 2172671. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. The gut’s feeling on bile acid signaling in NAFLD. Hepatobiliary Surg. Nutr. 2018, 7, 151–153. [Google Scholar] [CrossRef]
- Trauner, M.; Halilbasic, E. Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology 2011, 140, 1120–1125.e1–12. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, F.; Balas, I.; Robinson, M.J.; Bakdash, G. Border Control: The Role of the Microbiome in Regulating Epithelial Barrier Function. Cells 2024, 13, 477. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xu, Y.; Xia, Y.; Jia, X.; Chen, Y.; Liu, Y.; Zhang, L.; Chai, H.; Sun, L. Review on chronic metabolic diseases surrounding bile acids and gut microbiota: What we have explored so far. Life Sci. 2024, 336, 122304. [Google Scholar] [CrossRef]
- Slee, E.L.; McLennan, P.L.; Owen, A.J.; Theiss, M.L. Low dietary fish-oil threshold for myocardial membrane n-3 PUFA enrichment independent of n-6 PUFA intake in rats. J. Lipid Res. 2010, 51, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhai, X.; Wang, X.; Wu, Y.; Wang, H.; Qin, Y.; Han, J.; Meng, Y. Research progress on the relationship between bile acid metabolism and type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2023, 15, 235. [Google Scholar] [CrossRef]
- Lai, J.; Luo, L.; Zhou, T.; Feng, X.; Ye, J.; Zhong, B. Alterations in Circulating Bile Acids in Metabolic Dysfunction-Associated Steatotic Liver Disease: A Systematic Review and Meta-Analysis. Biomolecules 2023, 13, 1356. [Google Scholar] [CrossRef]
- Adams, L.A.; Wang, Z.; Liddle, C.; Melton, P.E.; Ariff, A.; Chandraratna, H.; Tan, J.; Ching, H.; Coulter, S.; de Boer, B.; et al. Bile acids associate with specific gut microbiota, low-level alcohol consumption and liver fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2020, 40, 1356–1365. [Google Scholar] [CrossRef]
- Kasai, Y.; Kessoku, T.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kobayashi, T.; Iwaki, M.; Ozaki, A.; Nogami, A.; Honda, Y.; et al. Association of Serum and Fecal Bile Acid Patterns With Liver Fibrosis in Biopsy-Proven Nonalcoholic Fatty Liver Disease: An Observational Study. Clin. Transl. Gastroenterol. 2022, 13, e00503. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.N.; Xu, C.F.; Liu, Y.R.; Sun, D.Q.; Jiang, L.; Tang, L.J.; Zhu, P.W.; Chen, S.D.; Liu, W.Y.; Wang, X.D.; et al. Secondary bile acids improve risk prediction for non-invasive identification of mild liver fibrosis in nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2023, 57, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Nimer, N.; Choucair, I.; Wang, Z.; Nemet, I.; Li, L.; Gukasyan, J.; Weeks, T.L.; Alkhouri, N.; Zein, N.; Tang, W.H.W.; et al. Bile acids profile, histopathological indices and genetic variants for non-alcoholic fatty liver disease progression. Metabolism 2021, 116, 154457. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Jiang, R.; Wang, X.; Liu, P.; Zhao, A.; Wu, Y.; Huang, F.; Liu, Z.; Rajani, C.; Zheng, X.; et al. Conjugated secondary 12α-hydroxylated bile acids promote liver fibrogenesis. eBioMedicine 2021, 66, 103290. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Sengupta, A.; Ricciotti, E.; Mrčela, A.; Mathew, D.; Mazaleuskaya, L.L.; Ghosh, S.; Brooks, T.G.; Turner, A.P.; Schanoski, A.S.; et al. Deep phenotyping of the lipidomic response in COVID-19 and non-COVID-19 sepsis. Clin. Transl. Med. 2023, 13, e1440. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease and cardiovascular risk: Metabolic aspects and novel treatments. Endocrine 2011, 40, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Scorletti, E.; Afolabi, P.R.; Miles, E.A.; Smith, D.E.; Almehmadi, A.; Alshathry, A.; Moyses, H.E.; Clough, G.F.; Wright, M.; Patel, J.; et al. Design and rationale of the INSYTE study: A randomised, placebo controlled study to test the efficacy of a synbiotic on liver fat, disease biomarkers and intestinal microbiota in non-alcoholic fatty liver disease. Contemp. Clin. Trials 2018, 71, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Eddowes, P.J.; Sasso, M.; Allison, M.; Tsochatzis, E.; Anstee, Q.M.; Sheridan, D.; Guha, I.N.; Cobbold, J.F.; Deeks, J.J.; Paradis, V.; et al. Accuracy of FibroScan Controlled Attenuation Parameter and Liver Stiffness Measurement in Assessing Steatosis and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhu, C.; Shao, L.; Ye, J.; Shen, Y.; Ren, Y. Role of Bile Acids in Dysbiosis and Treatment of Nonalcoholic Fatty Liver Disease. Mediat. Inflamm. 2019, 2019, 7659509. [Google Scholar] [CrossRef]
- Na, J.; Susan, S.B.; Adrian, C.-R.; Wensheng, L.; Colleen, A.N.; Maria, T.; Lucy, M.; Michael, J.B.; Robert, D.B.; Robert, J.G.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881. [Google Scholar]
- Gillard, J.; Clerbaux, L.-A.; Nachit, M.; Sempoux, C.; Staels, B.; Bindels, L.B.; Tailleux, A.; Leclercq, I.A. Bile acids contribute to the development of non-alcoholic steatohepatitis in mice. JHEP Rep. 2022, 4, 100387. [Google Scholar] [CrossRef] [PubMed]
- Clifford, B.L.; Sedgeman, L.R.; Williams, K.J.; Morand, P.; Cheng, A.; Jarrett, K.E.; Chan, A.P.; Brearley-Sholto, M.C.; Wahlström, A.; Ashby, J.W.; et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021, 33, 1671–1684.E4. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Discovery of farnesoid X receptor and its role in bile acid metabolism. Mol. Cell. Endocrinol. 2022, 548, 111618. [Google Scholar] [CrossRef] [PubMed]
- Barneda, D.; Planas-Iglesias, J.; Gaspar, M.L.; Mohammadyani, D.; Prasannan, S.; Dormann, D.; Han, G.S.; Jesch, S.A.; Carman, G.M.; Kagan, V.; et al. The brown adipocyte protein CIDEA promotes lipid droplet fusion via a phosphatidic acid-binding amphipathic helix. eLife 2015, 4, e07485. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Wang, J.; Li, Y.; Li, M.; Zhao, M.; Ge, K.; Zheng, D.; Cheung, K.C.P.; Liao, B.; Wang, S.; et al. Hyodeoxycholic acid alleviates non-alcoholic fatty liver disease through modulating the gut-liver axis. Cell Metab. 2023, 35, 1752–1766.e8. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Karrasch, T.; Schäffler, A. The emerging role of bile acids in white adipose tissue. Trends Endocrinol. Metab. 2023, 34, 718–734. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.; Othman, A.; Saely, C.H.; Rein, P.; Drexel, H.; von Eckardstein, A.; Rentsch, K.M. Bile acid metabolites in serum: Intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS ONE 2011, 6, e25006. [Google Scholar] [CrossRef]
- Li, W.; Shu, S.; Cheng, L.; Hao, X.; Wang, L.; Wu, Y.; Yuan, Z.; Zhou, J. Fasting serum total bile acid level is associated with coronary artery disease, myocardial infarction and severity of coronary lesions. Atherosclerosis 2020, 292, 193–200. [Google Scholar] [CrossRef]
- Wu, T.; Yang, M.; Xu, H.; Wang, L.; Wei, H.; Ji, G. Serum Bile Acid Profiles Improve Clinical Prediction of Nonalcoholic Fatty Liver in T2DM patients. J. Proteome Res. 2021, 20, 3814–3825. [Google Scholar] [CrossRef]
- Feng, X.; Zhai, G.; Yang, J.; Liu, Y.; Zhou, Y.; Guo, Q. Myocardial Infarction and Coronary Artery Disease in Menopausal Women With Type 2 Diabetes Mellitus Negatively Correlate With Total Serum Bile Acids. Front. Endocrinol. 2021, 12, 754006. [Google Scholar] [CrossRef]
- Chong Nguyen, C.; Duboc, D.; Rainteau, D.; Sokol, H.; Humbert, L.; Seksik, P.; Bellino, A.; Abdoul, H.; Bouazza, N.; Treluyer, J.-M.; et al. Circulating bile acids concentration is predictive of coronary artery disease in human. Sci. Rep. 2021, 11, 22661. [Google Scholar] [CrossRef]
- Mayerhofer, C.C.K.; Ueland, T.; Broch, K.; Vincent, R.P.; Cross, G.F.; Dahl, C.P.; Aukrust, P.; Gullestad, L.; Hov, J.R.; Trøseid, M. Increased Secondary/Primary Bile Acid Ratio in Chronic Heart Failure. J. Card. Fail. 2017, 23, 666–671. [Google Scholar] [CrossRef]
- Wen, W.; Li, Q.; She, J.; Bai, X.; Zhang, L.; Li, R.; Wu, Y.; Zhou, J.; Yuan, Z. Predictive value of serum TBA for 2-year MACEs in ACS patients undergoing PCI: A prospective cohort study. Sci. Rep. 2024, 14, 1733. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Fabregat, J.; Mostafa, H.; Sanchez-Gimenez, R.; Peiró, Ó.M.; Bonet, G.; Carrasquer, A.; Fragkiadakis, G.A.; Bardaji, A.; Bulló, M.; Papandreou, C. Bile Acids and Risk of Adverse Cardiovascular Events and All-Cause Mortality in Patients with Acute Coronary Syndrome. Nutrients 2024, 16, 1062. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. Linking liver metabolic and vascular disease via bile acid signaling. Trends Mol. Med. 2022, 28, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-D.; Xu, C.-F.; Chen, Q.-F.; Shapiro, M.D.; Lip, G.Y.H.; Chen, L.-L.; Targher, G.; Byrne, C.D.; Tian, N.; Xiao, T.; et al. Serum bile acid profiles are associated with heart failure with preserved ejection fraction in patients with metabolic dysfunction-associated fatty liver disease: An exploratory study. Diabetes Obes. Metab. 2024; early view. [Google Scholar]
- Marchianò, S.; Biagioli, M.; Bordoni, M.; Morretta, E.; Di Giorgio, C.; Vellecco, V.; Roselli, R.; Bellini, R.; Massa, C.; Cari, L.; et al. Defective Bile Acid Signaling Promotes Vascular Dysfunction, Supporting a Role for G-Protein Bile Acid Receptor 1/Farnesoid X Receptor Agonism and Statins in the Treatment of Nonalcoholic Fatty Liver Disease. J. Am. Heart Assoc. 2023, 12, e031241. [Google Scholar] [CrossRef]
- Marchisello, S.; Di Pino, A.; Scicali, R.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int. J. Mol. Sci. 2019, 20, 1948. [Google Scholar] [CrossRef]
- Desai, M.S.; Mathur, B.; Eblimit, Z.; Vasquez, H.; Taegtmeyer, H.; Karpen, S.J.; Penny, D.J.; Moore, D.D.; Anakk, S. Bile acid excess induces cardiomyopathy and metabolic dysfunctions in the heart. Hepatology 2017, 65, 189–201. [Google Scholar] [CrossRef]
- Mao, H.; Angelini, A.; Li, S.; Wang, G.; Li, L.; Patterson, C.; Pi, X.; Xie, L. CRAT links cholesterol metabolism to innate immune responses in the heart. Nat. Metab. 2023, 5, 1382–1394. [Google Scholar] [CrossRef]
- Yntema, T.; Koonen, D.P.Y.; Kuipers, F. Emerging Roles of Gut Microbial Modulation of Bile Acid Composition in the Etiology of Cardiovascular Diseases. Nutrients 2023, 15, 1850. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wei, J.; Yuan, H.; Li, Y.; Guo, Z. The role of the gut microbiota and bile acids in heart failure: A review. Medicine 2023, 102, e35795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Wu, W.; Zhu, Y.; Liu, X. The Role of Bile Acids in Cardiovascular Diseases: From Mechanisms to Clinical Implications. Aging Dis. 2023, 14, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, W.Q.; Fu, M.J.; Li, J.; Hu, C.H.; Chen, Y.; Zhou, M.M.; Gao, Z.J.; He, Y.L. Overview of bile acid signaling in the cardiovascular system. World J. Clin. Cases 2021, 9, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Ratziu, V.; Loomba, R.; Anstee, Q.M.; Kowdley, K.V.; Rinella, M.E.; Sheikh, M.Y.; Trotter, J.F.; Knapple, W.; Lawitz, E.J.; et al. Results from a new efficacy and safety analysis of the REGENERATE trial of obeticholic acid for treatment of pre-cirrhotic fibrosis due to non-alcoholic steatohepatitis. J. Hepatol. 2023, 79, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Taub, R.; Neff, G.W.; Lucas, K.J.; Labriola, D.; Moussa, S.E.; Alkhouri, N.; Bashir, M.R. Resmetirom for nonalcoholic fatty liver disease: A randomized, double-blind, placebo-controlled phase 3 trial. Nat. Med. 2023, 29, 2919–2928. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G.; Tilg, H. Thyroid hormone receptor-beta agonists: New MASLD therapies on the horizon. Gut 2024, 73, 573–581. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Zhu, N.L.; Huang, S.L.; Qu, H.; Gu, Y.P.; Qin, L.; Liu, J.; Leng, Y. A new mechanism of thyroid hormone receptor β agonists ameliorating nonalcoholic steatohepatitis by inhibiting intestinal lipid absorption via remodeling bile acid profiles. Acta Pharmacol. Sin. 2024. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Lopez, P.; Lawitz, E.J.; Lucas, K.J.; Loeffler, J.; Kim, W.; Goh, G.B.B.; Huang, J.F.; Serra, C.; Andreone, P.; et al. Tropifexor for nonalcoholic steatohepatitis: An adaptive, randomized, placebo-controlled phase 2a/b trial. Nat. Med. 2023, 29, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Liu, X.Y.; Zhan, W. Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des. Devel Ther. 2018, 12, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.1. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Gunn, N.; Neff, G.W.; Kohli, A.; Liu, L.; Flyer, A.; Goldkind, L.; Di Bisceglie, A.M. A phase 2, proof of concept, randomised controlled trial of berberine ursodeoxycholate in patients with presumed non-alcoholic steatohepatitis and type 2 diabetes. Nat. Commun. 2021, 12, 5503. [Google Scholar] [CrossRef] [PubMed]
- Jiao, T.Y.; Ma, Y.D.; Guo, X.Z.; Ye, Y.F.; Xie, C. Bile acid and receptors: Biology and drug discovery for nonalcoholic fatty liver disease. Acta Pharmacol. Sin. 2022, 43, 1103–1119. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.P. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef]
- Jahnel, J.; Zöhrer, E.; Alisi, A.; Ferrari, F.; Ceccarelli, S.; De Vito, R.; Scharnagl, H.; Stojakovic, T.; Fauler, G.; Trauner, M.; et al. Serum Bile Acid Levels in Children With Nonalcoholic Fatty Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 85–90. [Google Scholar] [CrossRef]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.-K.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, M.; Luo, Y.; Yang, W.; Yang, J.; Liu, J.; Zhou, J.; Xu, C.; Zhao, F.; Su, M.; et al. Ratio of Conjugated Chenodeoxycholic to Muricholic Acids is Associated with Severity of Nonalcoholic Steatohepatitis. Obesity 2019, 27, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Yara, S.; Ikegami, T.; Miyazaki, T.; Murakami, M.; Iwamoto, J.; Hirayama, T.; Kohjima, M.; Nakamuta, M.; Honda, A. Circulating bile acid profiles in Japanese patients with NASH. GastroHep 2019, 1, 302–310. [Google Scholar] [CrossRef]
- Caussy, C.; Hsu, C.; Singh, S. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment. Pharmacol. Ther. 2019, 49, 183–193. [Google Scholar] [CrossRef]
- Zhang, Z.; Dai, W.; Weng, S.; Luo, M.; Fu, J.; Zadroga, J.A.; Spolitu, S.; Peng, D. The association of serum total bile acid with non-alcoholic fatty liver disease in Chinese adults: A cross sectional study. Lipids Health Dis. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Sydor, S.; Best, J.; Messerschmidt, I.; Manka, P.; Vilchez-Vargas, R.; Brodesser, S.; Lucas, C.; Wegehaupt, A.; Wenning, C.; Aßmuth, S.; et al. Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin. Transl. Gastroenterol. 2020, 11, e00131. [Google Scholar] [CrossRef]
- Chen, T.; Zhou, K.; Sun, T.; Sang, C.; Jia, W.; Xie, G. Altered bile acid glycine: Taurine ratio in the progression of chronic liver disease. J. Gastroenterol. Hepatol. 2021, 37, 208–215. [Google Scholar] [CrossRef]
- Jung, Y.; Koo, B.K.; Jang, S.Y.; Kim, D.; Lee, H.; Lee, D.H.; Joo, S.K.; Jung, Y.J.; Park, J.H.; Yoo, T.; et al. Association between circulating bile acid alterations and nonalcoholic steatohepatitis independent of obesity and diabetes mellitus. Liver Int. 2021, 41, 2892–2902. [Google Scholar] [CrossRef]
- Rivera-Andrade, A.; Petrick, J.L.; Alvarez, C.S.; Graubard, B.I.; Florio, A.A.; Kroker-Lobos, M.F.; Parisi, D.; Freedman, N.D.; Lazo, M.; Guallar, E.; et al. Circulating bile acid concentrations and non-alcoholic fatty liver disease in Guatemala. Aliment. Pharmacol. Ther. 2022, 56, 321–329. [Google Scholar] [CrossRef]
- Fitzinger, J.; Rodriguez-Blanco, G.; Herrmann, M.; Borenich, A.; Stauber, R.; Aigner, E.; Mangge, H. Gender-Specific Bile Acid Profiles in Non-Alcoholic Fatty Liver Disease. Nutrients 2024, 16, 250. [Google Scholar] [CrossRef]
- Liu, T.-T.; Wang, J.; Liang, Y.; Wu, X.-Y.; Li, W.-Q.; Wang, Y.-H.; Jing, A.-R.; Liang, M.-M.; Sun, L.; Dou, J.; et al. The level of serum total bile acid is related to atherosclerotic lesions, prognosis and gut Lactobacillus in acute coronary syndrome patients. Ann. Med. 2023, 55, 2232369. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Variable | GCA | GCDCA | TCA | TCDCA | GDCA |
|---|---|---|---|---|---|
| Age (years) | −0.07 | −0.07 | −0.09 | −0.08 | 0.10 |
| Systolic blood pressure (mmHg) | −0.12 | −0.13 | −0.05 | −0.07 | 0.03 |
| Diastolic blood pressure (mmHg) | −0.11 | −0.10 | −0.01 | −0.05 | −0.10 |
| BMI (kg/m2) | 0.37 ** | 0.32 ** | 0.26 * | 0.26 * | 0.24 * |
| DEXA lean body mass (kg) | 0.05 | 0.02 | 0.05 | 0.02 | 0.03 |
| DEXA total body fat (%) | 0.23 * | 0.20 * | 0.11 | 0.11 | 0.19 |
| MRI SAT (%) † | 0.22 * | 0.15 | 0.14 | 0.12 | 0.18 |
| MRI VAT (%) † | −0.03 | 0.001 | −0.08 | −0.08 | 0.05 |
| Fasting glucose (mmol/L) † | 0.12 | 0.14 | 0.07 | 0.03 | 0.19 |
| Haemoglobin A1c (mmol/mol) † | 0.12 | 0.11 | 0.05 | 0.01 | 0.20 |
| Fasting insulin (mIU/L) † | 0.38 ** | 0.36 ** | 0.39 ** | 0.42 *** | 0.23 * |
| HOMA-IR † | 0.45 *** | 0.41 *** | 0.45 *** | 0.42 *** | 0.36 ** |
| AdipoIR † | 0.21 | 0.18 | 0.22 * | 0.25 * | 0.18 |
| Triglycerides (mmol/L) † | 0.18 | 0.15 | 0.21 * | 0.17 | 0.24 * |
| Total cholesterol (mmol/L) † | 0.04 | 0.01 | 0.08 | 0.11 | −0.02 |
| HDL cholesterol (mmol/L) | −0.09 | −0.08 | −0.05 | 0.001 | −0.01 |
| AST (IU/L) † | 0.31 * | 0.23 * | 0.33 ** | 0.33 ** | 0.29 * |
| ALT (IU/L) † | 0.24 * | 0.15 | 0.30 * | 0.27 * | 0.24 * |
| MRS-measured liver fat (%) † | 0.15 | 0.09 | 0.21 * | 0.20 * | 0.23 * |
| Liver VCTE (kPa) † | 0.39 *** | 0.32 * | 0.35 ** | 0.36 ** | 0.34 ** |
| FIB-4 index † | 0.21 * | 0.09 | 0.16 | 0.17 | 0.26 * |
| ELF test † | 0.09 | 0.05 | 0.11 | 0.12 | 0.16 |
| APRI index † | 0.26 * | 0.14 | 0.28 * | 0.25 | 0.25 * |
| Adiponectin (μg/mL) † | −0.19 | −0.13 | −0.25 | −0.18 | −0.06 |
| Leptin (ng/mL) † | 0.26 * | 0.20 | 0.16 | 0.16 | 0.25 * |
| TNFα (pg/mL) † | 0.29 * | 0.23 * | 0.33 * | 0.35 ** | 0.18 |
| IL-6 (pg/mL) † | 0.36 ** | 0.26 * | 0.33 * | 0.33 ** | 0.18 |
| IL-8 (pg/mL) † | 0.42 *** | 0.308 | 0.37 ** | 0.32 * | 0.35 ** |
| IL-10 (pg/mL) † | 0.28 * | 0.16 | 0.31 * | 0.30 * | 0.17 |
| hs-CRP (mg/L) † | 0.12 | 0.17 | 0.13 | 0.22 * | 0.03 |
| GDF-15 (pg/mL) | 0.24 * | 0.19 | 0.18 | 0.14 | 0.21 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilson, J.; Scorletti, E.; Swann, J.R.; Byrne, C.D. Bile Acids as Emerging Players at the Intersection of Steatotic Liver Disease and Cardiovascular Diseases. Biomolecules 2024, 14, 841. https://doi.org/10.3390/biom14070841
Bilson J, Scorletti E, Swann JR, Byrne CD. Bile Acids as Emerging Players at the Intersection of Steatotic Liver Disease and Cardiovascular Diseases. Biomolecules. 2024; 14(7):841. https://doi.org/10.3390/biom14070841
Chicago/Turabian StyleBilson, Josh, Eleonora Scorletti, Jonathan R. Swann, and Christopher D. Byrne. 2024. "Bile Acids as Emerging Players at the Intersection of Steatotic Liver Disease and Cardiovascular Diseases" Biomolecules 14, no. 7: 841. https://doi.org/10.3390/biom14070841