
Innate Immunity and MASLD

{kind=link}
Abstract
1. Introduction
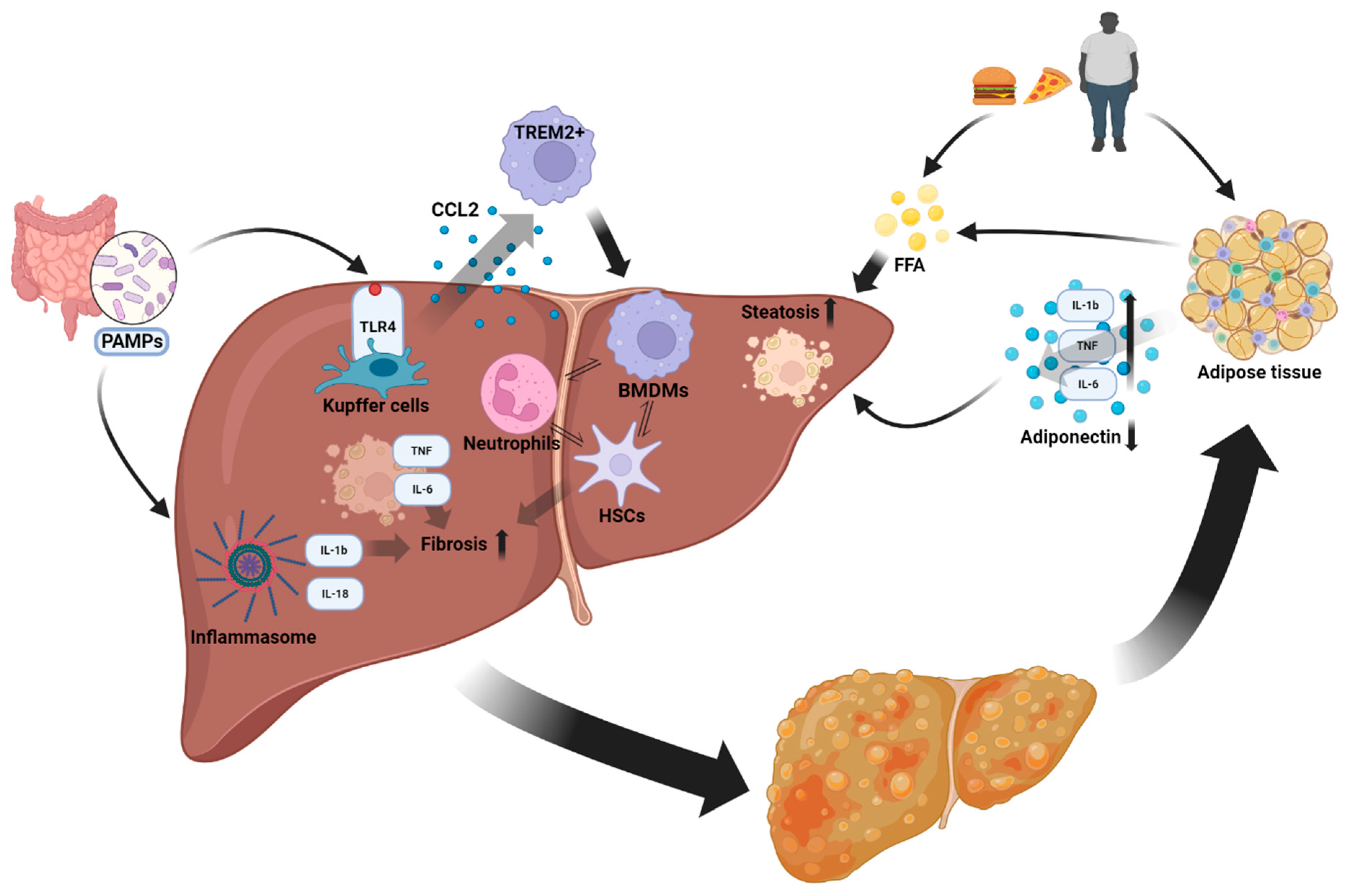
2. Proinflammatory Cytokines and Their Role in MASLD
3. Adipokines: Adipose Tissue-Derived Mediators Contributing to MASLD
4. Inflammasomes: Key Factors in MASLD
5. Various Cell Types Involved in Innate Immunity Contribute to This Disease
6. How Does Inflammation Evolve in MASLD?
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Targher, G.; Byrne, C.D.; Tilg, H. MASLD: A systemic metabolic disorder with cardiovascular and malignant complications. Gut 2024, 73, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P. NAFLD, obesity, and bariatric surgery. Gastroenterology 2006, 130, 1848–1852. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Tacke, F. Therapeutic modulation of the liver immune microenvironment. Hepatology 2023, 78, 1581–1601. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Chung, H.; Softic, S.; Moreno-Fernandez, M.E.; Divanovic, S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell Metab. 2023, 35, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Kotsiliti, E.; Leone, V.; Schuehle, S.; Govaere, O.; Li, H.; Wolf, M.J.; Horvatic, H.; Bierwirth, S.; Hundertmark, J.; Inverso, D.; et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J. Hepatol. 2023, 79, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Diehl, A.M. Cytokines in alcoholic and nonalcoholic steatohepatitis. N. Engl. J. Med. 2000, 343, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic fatty liver disease: Cytokine-adipokine interplay and regulation of insulin resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Wieckowska, A.; Papouchado, B.G.; Li, Z.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2008, 103, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Mawatari, H.; Fujita, K.; Iida, H.; Yonemitsu, K.; Kato, S.; Takahashi, H.; Kirikoshi, H.; Inamori, M.; Nozaki, Y.; et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J. Gastroenterol. 2007, 42, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamäki, J.; Kuulasmaa, T.; Kaminska, D.; Simonen, M.; Kärjä, V.; Grönlund, S.; Käkelä, P.; Pääkkönen, M.; Kainulainen, S.; Punnonen, K.; et al. Serum interleukin 1 receptor antagonist as an independent marker of non-alcoholic steatohepatitis in humans. J. Hepatol. 2012, 56, 663–670. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e310. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat. Metab. 2021, 3, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Barbuio, R.; Milanski, M.; Bertolo, M.B.; Saad, M.J.; Velloso, L.A. Infliximab reverses steatosis and improves insulin signal transduction in liver of rats fed a high-fat diet. J. Endocrinol. 2007, 194, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol. Metab. 2008, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.P.; Maachi, M.; Quignard-Boulangé, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef] [PubMed]
- Nov, O.; Shapiro, H.; Ovadia, H.; Tarnovscki, T.; Dvir, I.; Shemesh, E.; Kovsan, J.; Shelef, I.; Carmi, Y.; Voronov, E.; et al. Interleukin-1β regulates fat-liver crosstalk in obesity by auto-paracrine modulation of adipose tissue inflammation and expandability. PLoS ONE 2013, 8, e53626. [Google Scholar] [CrossRef]
- Negrin, K.A.; Roth Flach, R.J.; DiStefano, M.T.; Matevossian, A.; Friedline, R.H.; Jung, D.; Kim, J.K.; Czech, M.P. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis. PLoS ONE 2014, 9, e107265. [Google Scholar] [CrossRef]
- Moschen, A.R.; Molnar, C.; Enrich, B.; Geiger, S.; Ebenbichler, C.F.; Tilg, H. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol. Med. 2011, 17, 840–845. [Google Scholar] [CrossRef]
- Meier, C.A.; Bobbioni, E.; Gabay, C.; Assimacopoulos-Jeannet, F.; Golay, A.; Dayer, J.M. IL-1 receptor antagonist serum levels are increased in human obesity: A possible link to the resistance to leptin? J. Clin. Endocrinol. Metab. 2002, 87, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Dinarello, C.A. Suppression of inflammation and acquired immunity by IL-37. Immunol. Rev. 2018, 281, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Ballak, D.B.; van Diepen, J.A.; Moschen, A.R.; Jansen, H.J.; Hijmans, A.; Groenhof, G.J.; Leenders, F.; Bufler, P.; Boekschoten, M.V.; Müller, M.; et al. IL-37 protects against obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 4711. [Google Scholar] [CrossRef] [PubMed]
- Mountford, S.; Effenberger, M.; Noll-Puchta, H.; Griessmair, L.; Ringleb, A.; Haas, S.; Denk, G.; Reiter, F.P.; Mayr, D.; Dinarello, C.A.; et al. Modulation of Liver Inflammation and Fibrosis by Interleukin-37. Front. Immunol. 2021, 12, 603649. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Effenberger, M.; Adolph, T.E. A role for IL-1 inhibitors in the treatment of non-alcoholic fatty liver disease (NAFLD)? Expert Opin. Investig. Drugs 2020, 29, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Okubo, K.; Shimomura, I.; Funahashi, T.; Matsuzawa, Y.; Matsubara, K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem. Biophys. Res. Commun. 1996, 221, 286–289. [Google Scholar] [CrossRef]
- Hu, E.; Liang, P.; Spiegelman, B.M. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 1996, 271, 10697–10703. [Google Scholar] [CrossRef]
- Nakano, Y.; Tobe, T.; Choi-Miura, N.H.; Mazda, T.; Tomita, M. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J. Biochem. 1996, 120, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Terauchi, Y.; Yamauchi, T.; Kubota, T.; Moroi, M.; Matsui, J.; Eto, K.; Yamashita, T.; Kamon, J.; Satoh, H.; et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J. Biol. Chem. 2002, 277, 25863–25866. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med. 2002, 8, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Möhlig, M.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Adiponectin and protection against type 2 diabetes mellitus. Lancet 2003, 361, 226–228. [Google Scholar] [CrossRef]
- Kaser, S.; Moschen, A.; Kaser, A.; Ludwiczek, O.; Ebenbichler, C.F.; Vogel, W.; Jaschke, W.; Patsch, J.R.; Tilg, H. Circulating adiponectin reflects severity of liver disease but not insulin sensitivity in liver cirrhosis. J. Intern. Med. 2005, 258, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Enrich, B.; Tilg, H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem. Biophys. Res. Commun. 2004, 323, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.F.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Molnar, C.; Wolf, A.M.; Weiss, H.; Graziadei, I.; Kaser, S.; Ebenbichler, C.F.; Stadlmann, S.; Moser, P.L.; Tilg, H. Effects of weight loss induced by bariatric surgery on hepatic adipocytokine expression. J. Hepatol. 2009, 51, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schönauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects With Non-alcoholic Fatty Liver. Am. J. Gastroenterol. 2017, 112, 102–110. [Google Scholar] [CrossRef]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Membrez, M.; Ammon-Zufferey, C.; Philippe, D.; Aprikian, O.; Monnard, I.; Macé, K.; Darimont, C. Interleukin-18 protein level is upregulated in adipose tissue of obese mice. Obesity (Silver Spring) 2009, 17, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Kui, L.; Reca, A.; Leszczynska, A.; Kim, A.D.; Booshehri, L.M.; Wree, A.; Friess, H.; Hartmann, D.; Broderick, L.; et al. Cell-specific Deletion of NLRP3 Inflammasome Identifies Myeloid Cells as Key Drivers of Liver Inflammation and Fibrosis in Murine Steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Inzaugarat, M.E.; Johnson, C.D.; Holtmann, T.M.; McGeough, M.D.; Trautwein, C.; Papouchado, B.G.; Schwabe, R.; Hoffman, H.M.; Wree, A.; Feldstein, A.E. NLR Family Pyrin Domain-Containing 3 Inflammasome Activation in Hepatic Stellate Cells Induces Liver Fibrosis in Mice. Hepatology 2019, 69, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Quezada, N.; Valencia, I.; Torres, J.; Maturana, G.; Cerda, J.; Arab, J.P.; Fuentes, J.J.; Pinto, C.; Turiel, D.; Cortés, V. Insulin resistance and liver histopathology in metabolically unhealthy subjects do not correlate with the hepatic abundance of NLRP3 inflammasome nor circulating IL-1β levels. BMJ Open Diabetes Res. Care 2021, 9, e001975. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Horn, C.L.; Kothari, V.; Yeh, M.M.; Shyu, I.; Lee, S.P.; Savard, C.E. Genetic deletion or pharmacologic inhibition of the Nlrp3 inflammasome did not ameliorate experimental NASH. J. Lipid Res. 2023, 64, 100330. [Google Scholar] [CrossRef]
- Liao, C.Y.; Song, M.J.; Gao, Y.; Mauer, A.S.; Revzin, A.; Malhi, H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front. Immunol. 2018, 9, 2980. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.L.; Vorac, J.; Hofmann, K.; Meiser, P.; Unterweger, I.; Kuerschner, L.; Weighardt, H.; Förster, I.; Thiele, C. Kupffer Cells Sense Free Fatty Acids and Regulate Hepatic Lipid Metabolism in High-Fat Diet and Inflammation. Cells 2020, 9, 2258. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Vespasiani-Gentilucci, U.; Carotti, S.; Perrone, G.; Mazzarelli, C.; Galati, G.; Onetti-Muda, A.; Picardi, A.; Morini, S. Hepatic toll-like receptor 4 expression is associated with portal inflammation and fibrosis in patients with NAFLD. Liver Int. 2015, 35, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Blériot, C.; Barreby, E.; Dunsmore, G.; Ballaire, R.; Chakarov, S.; Ficht, X.; De Simone, G.; Andreata, F.; Fumagalli, V.; Guo, W.; et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 2021, 54, 2101–2116.e2106. [Google Scholar] [CrossRef]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e614. [Google Scholar] [CrossRef]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e1057. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Baba, I.; Poupel, L.; Dussaud, S.; Moreau, M.; Gélineau, A.; Marcelin, G.; Magréau-Davy, E.; Ouhachi, M.; Lesnik, P.; et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 2020, 53, 627–640.e625. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Kato, H.; Suganami, T.; Konuma, K.; Marumoto, Y.; Terai, S.; Sakugawa, H.; Kanai, S.; Hamaguchi, M.; Fukaishi, T.; et al. Hepatic crown-like structure: A unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS ONE 2013, 8, e82163. [Google Scholar] [CrossRef] [PubMed]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e645. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Q.; Zhou, C.; Xu, Y.; Liu, D.; Fujiwara, N.; Kubota, N.; Click, A.; Henderson, P.; Vancil, J.; et al. Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity 2023, 56, 58–77.e11. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Porsch, F.; Kiss, M.G.; Rajcic, D.; Papac-Miličević, N.; Hoebinger, C.; Goederle, L.; Hladik, A.; Shaw, L.E.; Horstmann, H.; et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J. Hepatol. 2022, 77, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Boesch, M.; Lindhorst, A.; Feio-Azevedo, R.; Brescia, P.; Silvestri, A.; Lannoo, M.; Deleus, E.; Jaekers, J.; Topal, H.; Topal, B.; et al. Adipose tissue macrophage dysfunction is associated with a breach of vascular integrity in NASH. J. Hepatol. 2024, 80, 397–408. [Google Scholar] [CrossRef]
- Ye, D.; Yang, K.; Zang, S.; Lin, Z.; Chau, H.T.; Wang, Y.; Zhang, J.; Shi, J.; Xu, A.; Lin, S.; et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 2016, 65, 988–997. [Google Scholar] [CrossRef]
- Gao, B.; Tsukamoto, H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology 2016, 150, 1704–1709. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, L.; Chang, N.; Hou, L.; Zhou, X.; Yang, L.; Li, L. Neutrophils undergo switch of apoptosis to NETosis during murine fatty liver injury via S1P receptor 2 signaling. Cell Death Dis. 2020, 11, 379. [Google Scholar] [CrossRef]
- van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O‘Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef]
- Hwang, S.; He, Y.; Xiang, X.; Seo, W.; Kim, S.J.; Ma, J.; Ren, T.; Park, S.H.; Zhou, Z.; Feng, D.; et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology 2020, 72, 412–429. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Cai, Y.; Wang, W.; Jiang, J.X.; Varga, Z.V.; Feng, D.; Pacher, P.; Kunos, G.; Torok, N.J.; et al. Neutrophil-Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 2018, 5, 399–413. [Google Scholar] [CrossRef]
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482. [Google Scholar] [CrossRef]
- Alkhouri, N.; Morris-Stiff, G.; Campbell, C.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Zein, N.N.; Feldstein, A.E. Neutrophil to lymphocyte ratio: A new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int. 2012, 32, 297–302. [Google Scholar] [CrossRef]
- Calvente, C.J.; Tameda, M.; Johnson, C.D.; Del Pilar, H.; Lin, Y.C.; Adronikou, N.; De Mollerat Du Jeu, X.; Llorente, C.; Boyer, J.; Feldstein, A.E. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J. Clin. Investig. 2019, 129, 4091–4109. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef]
- Leinwand, J.C.; Paul, B.; Chen, R.; Xu, F.; Sierra, M.A.; Paluru, M.M.; Nanduri, S.; Alcantara, C.G.; Shadaloey, S.A.; Yang, F.; et al. Intrahepatic microbes govern liver immunity by programming NKT cells. J. Clin. Investig. 2022, 132, e151725. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y.; Tsuneyama, K.; Sugiyama, T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 673–680. [Google Scholar] [CrossRef]
- Adler, M.; Taylor, S.; Okebugwu, K.; Yee, H.; Fielding, C.; Fielding, G.; Poles, M. Intrahepatic natural killer T cell populations are increased in human hepatic steatosis. World J. Gastroenterol. 2011, 17, 1725–1731. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Cuff, A.O.; Sillito, F.; Dertschnig, S.; Hall, A.; Luong, T.V.; Chakraverty, R.; Male, V. The Obese Liver Environment Mediates Conversion of NK Cells to a Less Cytotoxic ILC1-Like Phenotype. Front. Immunol. 2019, 10, 2180. [Google Scholar] [CrossRef]
- Amer, J.; Salhab, A.; Noureddin, M.; Doron, S.; Abu-Tair, L.; Ghantous, R.; Mahamid, M.; Safadi, R. Insulin signaling as a potential natural killer cell checkpoint in fatty liver disease. Hepatol. Commun. 2018, 2, 285–298. [Google Scholar] [CrossRef]
- Gur, C.; Doron, S.; Kfir-Erenfeld, S.; Horwitz, E.; Abu-Tair, L.; Safadi, R.; Mandelboim, O. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut 2012, 61, 885–893. [Google Scholar] [CrossRef]
- Melhem, A.; Muhanna, N.; Bishara, A.; Alvarez, C.E.; Ilan, Y.; Bishara, T.; Horani, A.; Nassar, M.; Friedman, S.L.; Safadi, R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 2006, 45, 60–71. [Google Scholar] [CrossRef]
- Muhanna, N.; Abu Tair, L.; Doron, S.; Amer, J.; Azzeh, M.; Mahamid, M.; Friedman, S.; Safadi, R. Amelioration of hepatic fibrosis by NK cell activation. Gut 2011, 60, 90–98. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, X.; Liu, W.; Zhou, Y.; Wei, W.; Liu, D.; Wong, C.C.; Sung, J.J.Y.; Yu, J. Activated Natural Killer Cell Promotes Nonalcoholic Steatohepatitis Through Mediating JAK/STAT Pathway. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 257–274. [Google Scholar] [CrossRef]
- Kahraman, A.; Schlattjan, M.; Kocabayoglu, P.; Yildiz-Meziletoglu, S.; Schlensak, M.; Fingas, C.D.; Wedemeyer, I.; Marquitan, G.; Gieseler, R.K.; Baba, H.A.; et al. Major histocompatibility complex class I-related chains A and B (MIC A/B): A novel role in nonalcoholic steatohepatitis. Hepatology 2010, 51, 92–102. [Google Scholar] [CrossRef]
- Martínez-Chantar, M.L.; Delgado, T.C.; Beraza, N. Revisiting the Role of Natural Killer Cells in Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2021, 12, 640869. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Sookoian, S.; Salatino, A.; Castaño, G.O.; Landa, M.S.; Fijalkowky, C.; Garaycoechea, M.; Pirola, C.J. Intrahepatic bacterial metataxonomic signature in non-alcoholic fatty liver disease. Gut 2020, 69, 1483–1491. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E. Liver microbes controlling immunity: Facts and pitfalls. Cell Metab. 2022, 34, 510–512. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, M.; Schwärzler, J.; Jukic, A.; Tilg, H. Innate Immunity and MASLD. Biomolecules 2024, 14, 476. https://doi.org/10.3390/biom14040476
Meyer M, Schwärzler J, Jukic A, Tilg H. Innate Immunity and MASLD. Biomolecules. 2024; 14(4):476. https://doi.org/10.3390/biom14040476
Chicago/Turabian StyleMeyer, Moritz, Julian Schwärzler, Almina Jukic, and Herbert Tilg. 2024. "Innate Immunity and MASLD" Biomolecules 14, no. 4: 476. https://doi.org/10.3390/biom14040476
APA StyleMeyer, M., Schwärzler, J., Jukic, A., & Tilg, H. (2024). Innate Immunity and MASLD. Biomolecules, 14(4), 476. https://doi.org/10.3390/biom14040476