
Regulation of Fructose Metabolism in Nonalcoholic Fatty Liver Disease
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fructose Metabolism and Metabolic Tissues
2.1. Overview of Fructose Metabolism
2.2. Fructose and Intestine
2.3. Fructose and Liver
2.4. Fructose and Adipose Tissue
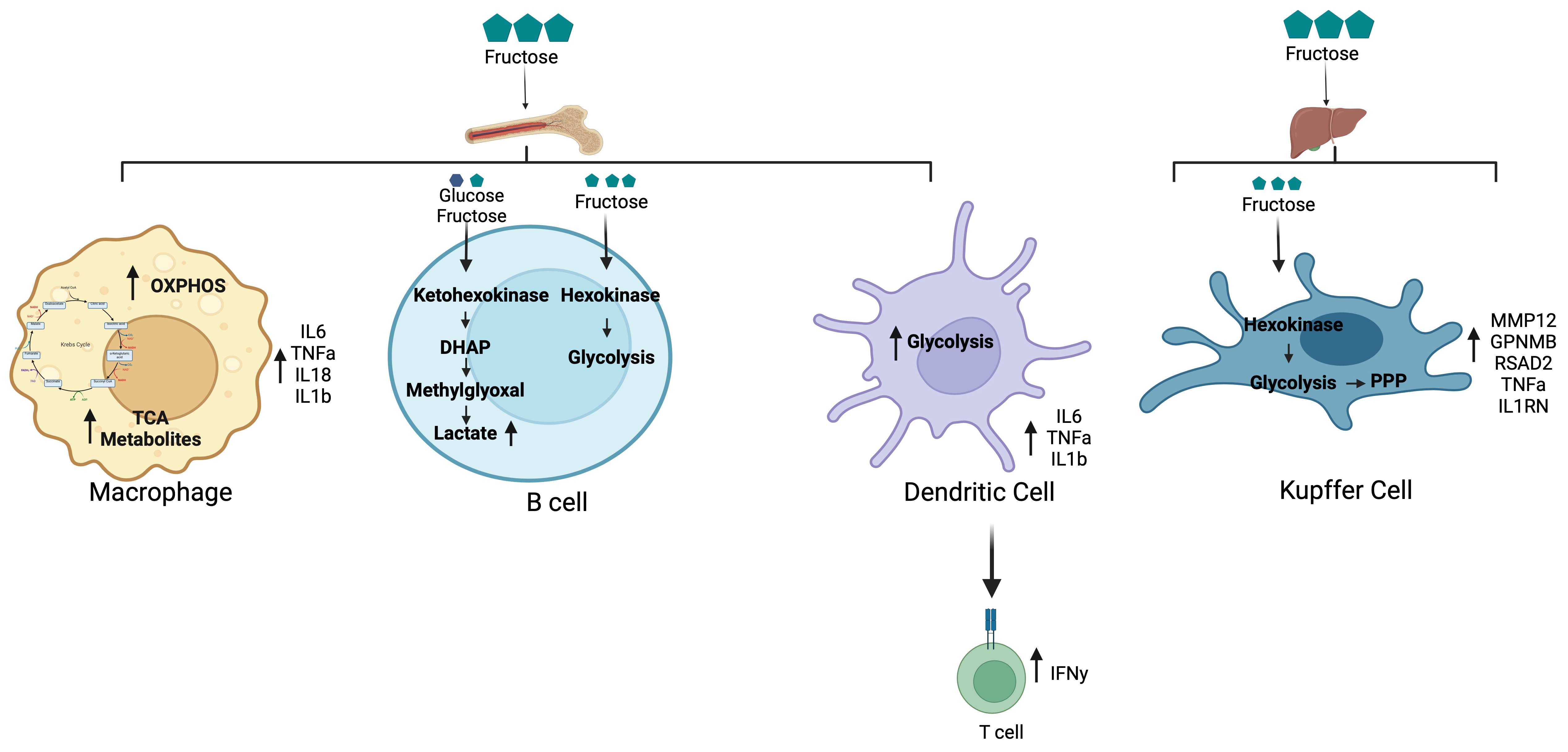
2.5. Fructose Metabolism Regulation of Immune Cell Function
2.5.1. Myeloid Cells
2.5.2. B Cells
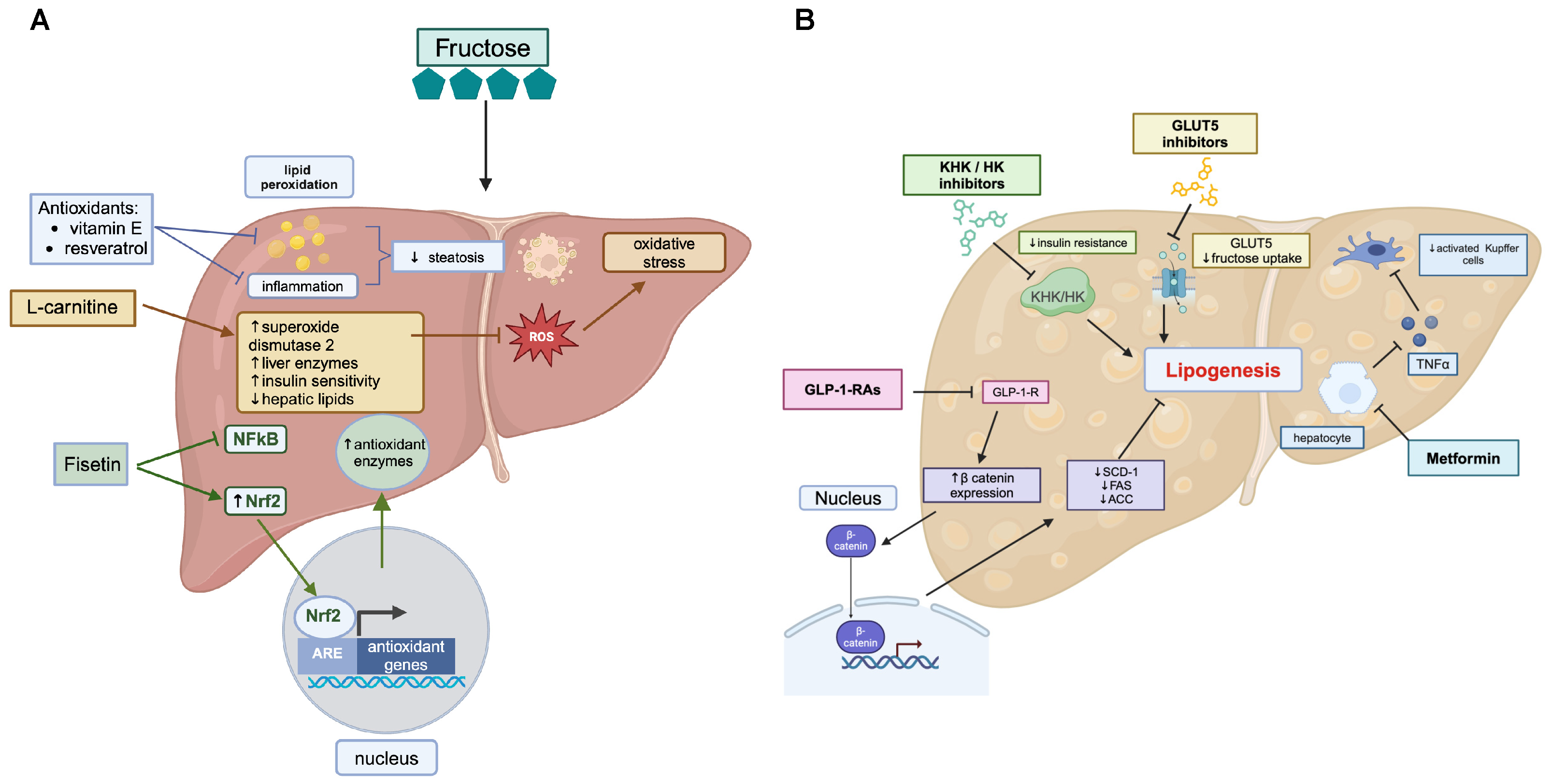
3. Regulation of Fructose Intake and Metabolism to Prevent Metabolic Disease
3.1. Dietary Modifications and Physical Activity
3.2. Nutritional Supplements
3.3. Pharmacological Interventions
3.4. Fructose Transporter Modulators
3.5. Insulin-Sensitizing Agents
4. Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, M.; Gastaldelli, A.; Hyötyläinen, T.; Arretxe, E.; Alonso, C.; Gaggini, M.; Brosnan, J.; Anstee, Q.M.; Millet, O.; Ortiz, P.; et al. Metabolomics and lipidomics in NAFLD: Biomarkers and non-invasive diagnostic tests. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 835–856. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J.; Association, A.G. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [PubMed]
- Kneeman, J.M.; Misdraji, J.; Corey, K.E. Secondary causes of nonalcoholic fatty liver disease. Therap Adv. Gastroenterol. 2012, 5, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.; Kennedy, L.; Hargrove, L.; Demieville, J.; Thomson, J.; Alpini, G.; Francis, H. Updates on Dietary Models of Nonalcoholic Fatty Liver Disease: Current Studies and Insights. Gene Expr. 2018, 18, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Popescu, M.; Popescu, I.A.; Stanciu, M.; Cazacu, S.M.; Ianosi, N.G.; Comanescu, M.V.; Singer, C.E.; Neagoe, C.D. Non-alcoholic fatty liver disease—Clinical and histopathological aspects. Rom. J. Morphol. Embryol. 2016, 57, 1295–1302. [Google Scholar] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed]
- Önnerhag, K.; Nilsson, P.M.; Lindgren, S. Increased risk of cirrhosis and hepatocellular cancer during long-term follow-up of patients with biopsy-proven NAFLD. Scand. J. Gastroenterol. 2014, 49, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Cooksley, W.G.; Hanson, R.; Searle, J.; Halliday, J.W.; Powell, L.W. The natural history of nonalcoholic steatohepatitis: A follow-up study of forty-two patients for up to 21 years. Hepatology 1990, 11, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology 1999, 29, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla Bertot, L.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol 2011, 9, 524–530.e1. quiz e560. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.M.; Price, J.F.; Glancy, S.; Perry, E.; Nee, L.D.; Hayes, P.C.; Frier, B.M.; Van Look, L.A.; Johnston, G.I.; Reynolds, R.M.; et al. Prevalence of and risk factors for hepatic steatosis and nonalcoholic Fatty liver disease in people with type 2 diabetes: The Edinburgh Type 2 Diabetes Study. Diabetes Care 2011, 34, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Muzurovic, E.; Mikhailidis, D.P.; Mantzoros, C. Non-alcoholic fatty liver disease, insulin resistance, metabolic syndrome and their association with vascular risk. Metabolism 2021, 119, 154770. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kwon, Y.J.; Park, K.; Lee, H.S.; Park, H.K.; Han, J.H.; Ahn, S.B. Metabolic Score for Insulin Resistance Is Inversely Related to Incident Advanced Liver Fibrosis in Patients with Non-Alcoholic Fatty Liver Disease. Nutrients 2022, 14, 3039. [Google Scholar] [CrossRef] [PubMed]
- Chew, N.W.S.; Hui Pan, X.; Chong, B.; Chandramouli, C.; Muthiah, M.; Lam, C.S.P. Type 2 diabetes mellitus and cardiometabolic outcomes in metabolic dysfunction-associated steatotic liver disease population. Diabetes Res. Clin. Pract. 2024, 211, 111652. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nakagawa, T.; Taniguchi, H.; Fujii, K.; Omatsu, T.; Nakajima, T.; Sarui, H.; Shimazaki, M.; et al. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann. Intern. Med. 2005, 143, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Hillenbrand, A.; Kiebler, B.; Schwab, C.; Scheja, L.; Xu, P.; Henne-Bruns, D.; Wolf, A.M.; Knippschild, U. Prevalence of non-alcoholic fatty liver disease in four different weight related patient groups: Association with small bowel length and risk factors. BMC Res. Notes 2015, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bedossa, P.; Guy, C.D.; Schattenberg, J.M.; Loomba, R.; Taub, R.; Labriola, D.; Moussa, S.E.; Neff, G.W.; Rinella, M.E.; et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N. Engl. J. Med. 2024, 390, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, M.; Yamasandhi, P.G. Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, Z.; Whiting, S.J.; Vatanparast, H. Current evidence on the association of the metabolic syndrome and dietary patterns in a global perspective. Nutr. Res. Rev. 2016, 29, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1228S–1235S. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.T.; Chan, T.F.; Huang, H.L.; Lee, C.Y.; Tsai, S.; Wu, P.W.; Yang, Y.C.; Wang, T.N.; Lee, C.H. Fructose-Rich Beverage Intake and Central Adiposity, Uric Acid, and Pediatric Insulin Resistance. J. Pediatr. 2016, 171, 90–96.e1. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, L.R. 40 years of adding more fructose to high fructose corn syrup than is safe, through the lens of malabsorption and altered gut health-gateways to chronic disease. Nutr. J. 2024, 23, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Luan, Y.; Li, Y.; Ye, S.; Wang, G.; Cai, X.; Liang, Y.; Kord Varkaneh, H.; Luan, Y. The effect of high-fructose corn syrup vs. sucrose on anthropometric and metabolic parameters: A systematic review and meta-analysis. Front. Nutr. 2022, 9, 1013310. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Sucrose, high-fructose corn syrup, and fructose, their metabolism and potential health effects: What do we really know? Adv. Nutr. 2013, 4, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef] [PubMed]
- Faller, J.; Fox, I.H. Ethanol-induced hyperuricemia: Evidence for increased urate production by activation of adenine nucleotide turnover. N. Engl. J. Med. 1982, 307, 1598–1602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, H.; Cheng, X.; Li, Y.; Zhao, Y.; Mei, W.; Wei, X.; Zhou, H.; Du, Y.; Zeng, C. Dietary intake of fructose increases purine de novo synthesis: A crucial mechanism for hyperuricemia. Front. Nutr. 2022, 9, 1045805. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Harman, J.L.; Coresh, J.; Kottgen, A.; McAdams-DeMarco, M.A.; Correa, A.; Young, B.A.; Katz, R.; Rebholz, C.M. The Dietary Fructose:Vitamin C Intake Ratio Is Associated with Hyperuricemia in African-American Adults. J. Nutr. 2018, 148, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, S.; Xu, W.; Zhang, J.; Fu, W.; Feng, C. Association between the hyperuricemia and nonalcoholic fatty liver disease risk in a Chinese population: A retrospective cohort study. PLoS ONE 2017, 12, e0177249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yang, N.; Xing, X.; Chang, D.; Li, J.; Deng, J.; Chen, Y.; Hu, C.; Zhang, R.; Lu, X.; et al. Obesity interacts with hyperuricemia on the severity of non-alcoholic fatty liver disease. BMC Gastroenterol. 2021, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liang, Z.; Ma, J.; Hu, D.; Yao, F.; Qin, P. Total sugar, added sugar, fructose, and sucrose intake and all-cause, cardiovascular, and cancer mortality: A systematic review and dose-response meta-analysis of prospective cohort studies. Nutrition 2023, 111, 112032. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S.M.; Kaiser, A.; Eichner, G.; Fasshauer, M. Association of sugar intake from different sources with cardiovascular disease incidence in the prospective cohort of UK Biobank participants. Nutr. J. 2024, 23, 22. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Tayyiba, M.; Agarwal, A.; Mejia, S.B.; de Souza, R.J.; Wolever, T.M.S.; Leiter, L.A.; Kendall, C.W.C.; Jenkins, D.J.A.; Sievenpiper, J.L. Relation of Total Sugars, Sucrose, Fructose, and Added Sugars with the Risk of Cardiovascular Disease: A Systematic Review and Dose-Response Meta-analysis of Prospective Cohort Studies. Mayo Clin. Proc. 2019, 94, 2399–2414. [Google Scholar] [CrossRef] [PubMed]
- Kilonzo, S.B.; Kamala, E.; Jaka, H.; Ngoya, P. Non-alcoholic fatty liver disease in Tanzania: Prevalence, determinants, and diagnostic performance of triglycerides-glucose index and triglycerides-glucose inde -body mass index compared to the hepatic ultrasound in overweight and obese individuals. BMC Gastroenterol. 2024, 24, 96. [Google Scholar] [CrossRef] [PubMed]
- Wahjudi, P.N.; Patterson, M.E.; Lim, S.; Yee, J.K.; Mao, C.S.; Lee, W.N. Measurement of glucose and fructose in clinical samples using gas chromatography/mass spectrometry. Clin. Biochem. 2010, 43, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Calle, R.A. Elevated Serum Sorbitol and not Fructose in Type 2 Diabetic Patients. Biomark. Insights 2010, 5, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.O.; Hausman, A.M.; Ifkovits, C.A.; Buse, J.B.; Gould, G.W.; Burant, C.F.; Bell, G.I. Human intestinal glucose transporter expression and localization of GLUT5. Am. J. Physiol. 1992, 262, C795–C800. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, R.P. Dietary and developmental regulation of intestinal sugar transport. Biochem. J. 2001, 360, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Kretowicz, M.; Johnson, R.J.; Ishimoto, T.; Nakagawa, T.; Manitius, J. The impact of fructose on renal function and blood pressure. Int. J. Nephrol. 2011, 2011, 315879. [Google Scholar] [CrossRef] [PubMed]
- Dyer, J.; Wood, I.S.; Palejwala, A.; Ellis, A.; Shirazi-Beechey, S.P. Expression of monosaccharide transporters in intestine of diabetic humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G241–G248. [Google Scholar] [CrossRef] [PubMed]
- Stuart, C.A.; Howell, M.E.; Yin, D. Overexpression of GLUT5 in diabetic muscle is reversed by pioglitazone. Diabetes Care 2007, 30, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, J.; Wlodarczyk, M.; Zielinska, M.; Jedrzejczak, B.; Dziki, L.; Fichna, J. Blockade of fructose transporter protein GLUT5 inhibits proliferation of colon cancer cells: Proof of concept for a new class of anti-tumor therapeutics. Pharmacol. Rep. 2021, 73, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Wuest, M.; Hamann, I.; Bouvet, V.; Glubrecht, D.; Marshall, A.; Trayner, B.; Soueidan, O.M.; Krys, D.; Wagner, M.; Cheeseman, C.; et al. Molecular Imaging of GLUT1 and GLUT5 in Breast Cancer: A Multitracer Positron Emission Tomography Imaging Study in Mice. Mol. Pharmacol. 2018, 93, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Maianu, L.; Melbert, B.R.; Garvey, W.T. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: A role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol. Dis. 2004, 32, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Malide, D.; Davies-Hill, T.M.; Levine, M.; Simpson, I.A. Distinct localization of GLUT-1, -3, and -5 in human monocyte-derived macrophages: Effects of cell activation. Am. J. Physiol. 1998, 274, E516–E526. [Google Scholar] [CrossRef] [PubMed]
- Grossbard, L.; Schimke, R.T. Multiple hexokinases of rat tissues. Purification and comparison of soluble forms. J. Biol. Chem. 1966, 241, 3546–3560. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, X.; Peng, L.X.; Jiang, Y.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Lee, J.H.; Cote, G.; Wang, H.; et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 2016, 18, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kang, J.; Kang, Y.L.; Woo, J.; Kim, Y.; Huh, J.; Park, J.W. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 2020, 11, 5436. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, T.; Schonenberger, M.J.; Walter, K.M.; Charles, K.N.; Faust, P.L.; Kovacs, W.J. Peroxisome-Deficiency and HIF-2alpha Signaling Are Negative Regulators of Ketohexokinase Expression. Front. Cell Dev. Biol. 2020, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [PubMed]
- Debosch, B.J.; Chen, Z.; Saben, J.L.; Finck, B.N.; Moley, K.H. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J. Biol. Chem. 2014, 289, 10989–10998. [Google Scholar] [CrossRef] [PubMed]
- Novelle, M.G.; Bravo, S.B.; Deshons, M.; Iglesias, C.; García-Vence, M.; Annells, R.; da Silva Lima, N.; Nogueiras, R.; Fernández-Rojo, M.A.; Diéguez, C.; et al. Impact of liver-specific GLUT8 silencing on fructose-induced inflammation and omega oxidation. iScience 2021, 24, 102071. [Google Scholar] [CrossRef] [PubMed]
- Colville, C.A.; Seatter, M.J.; Jess, T.J.; Gould, G.W.; Thomas, H.M. Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: Substrate specificities and effects of transport inhibitors. Biochem. J. 1993, 290, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Oppelt, S.A.; Sennott, E.M.; Tolan, D.R. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol. Genet. Metab. 2015, 114, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, T.; Liao, Y.; Wang, Y.; Gao, Y.; Hu, H.; Huang, H.; Wu, F.; Chen, Y.G.; Xu, S.; et al. Triose Kinase Controls the Lipogenic Potential of Fructose and Dietary Tolerance. Cell Metab. 2020, 32, 605–618.e7. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, O.; Felig, P. Role of the kidney in the metabolism of fructose in 60-hour fasted humans. Diabetes 1982, 31, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Lodge, M.; Scheidemantle, G.; Adams, V.R.; Cottam, M.A.; Richard, D.; Breuer, D.; Thompson, P.; Shrestha, K.; Liu, X.; Kennedy, A. Fructose regulates the pentose phosphate pathway and induces an inflammatory and resolution phenotype in Kupffer cells. Sci. Rep. 2024, 14, 4020. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Blagih, J.; Zani, F.; Rees, A.; Hill, D.G.; Jenkins, B.J.; Bull, C.J.; Moreira, D.; Bantan, A.I.M.; Cronin, J.G.; et al. Fructose reprogrammes glutamine-dependent oxidative metabolism to support LPS-induced inflammation. Nat. Commun. 2021, 12, 1209. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, T.; Du, M.; He, S.; Huang, N.; Cheng, B.; Yan, C.; Tang, W.; Gao, W.; Guo, H.; et al. Ketohexokinase-dependent metabolism of cerebral endogenous fructose in microglia drives diabetes-associated cognitive dysfunction. Exp. Mol. Med. 2023, 55, 2417–2432. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Maeno, S.; Tanizawa, Y.; Kneifel, W.; Arita, M.; Dicks, L.; Salminen, S. Fructophilic Lactic Acid Bacteria, a Unique Group of Fructose-Fermenting Microbes. Appl. Environ. Microbiol. 2018, 84, 19. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.L.; Kim, J.; Kwak, S.B.; Kim, Y.S.; Huh, J.; Park, J.W. The polyol pathway and nuclear ketohexokinase A signaling drive hyperglycemia-induced metastasis of gastric cancer. Exp. Mol. Med. 2024, 56, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Cicerchi, C.; Pedler, M.; Petrash, M.J.; Johnson, R.J.; Tolan, D.R.; Lanaspa, M.A. Endogenous Fructose Production and Metabolism Drive Metabolic Dysregulation and Liver Disease in Mice with Hereditary Fructose Intolerance. Nutrients 2023, 15, 4376. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Choi, W.M.; Kim, J.S.; Jung, Y.; Lee, S.Y.; Seo, H.R.; Kim, K.M. Serum Sorbitol Dehydrogenase as a Novel Prognostic Factor for Hepatocellular Carcinoma after Surgical Resection. Cancers 2021, 13, 6143. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.R.; Ramsamooj, S.; Liang, R.J.; Katti, A.; Pozovskiy, R.; Vasan, N.; Hwang, S.K.; Nahiyaan, N.; Francoeur, N.J.; Schatoff, E.M.; et al. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 2021, 597, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Dong, H.; Chen, Z.; Jin, M.; Yin, J.; Li, H.; Shi, D.; Shao, Y.; Wang, H.; Chen, T.; et al. Intestinal Microbiota Mediates High-Fructose and High-Fat Diets to Induce Chronic Intestinal Inflammation. Front. Cell Infect. Microbiol. 2021, 11, 654074. [Google Scholar] [CrossRef] [PubMed]
- Montrose, D.C.; Nishiguchi, R.; Basu, S.; Staab, H.A.; Zhou, X.K.; Wang, H.; Meng, L.; Johncilla, M.; Cubillos-Ruiz, J.R.; Morales, D.K.; et al. Dietary Fructose Alters the Composition, Localization, and Metabolism of Gut Microbiota in Association with Worsening Colitis. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 525–550. [Google Scholar] [CrossRef] [PubMed]
- Letsinger, A.C.; Menon, R.; Iyer, A.R.; Vellers, H.L.; Granados, J.Z.; Jayaraman, A.; Lightfoot, J.T. A High Fat/High Sugar Diet Alters the Gastrointestinal Metabolome in a Sex Dependent Manner. Metabolites 2020, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Ham, M.; Lee, J.W.; Choi, A.H.; Jang, H.; Choi, G.; Park, J.; Kozuka, C.; Sears, D.D.; Masuzaki, H.; Kim, J.B. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol. Cell Biol. 2013, 33, 2425–2435. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, L.; Li, X.; Hao, R.; Li, J. Effects of high fructose corn syrup on intestinal microbiota structure and obesity in mice. NPJ Sci. Food 2022, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Alexander, L.M.; Pan, M.; Schueler, K.L.; Keller, M.P.; Attie, A.D.; Walter, J.; van Pijkeren, J.P. Dietary Fructose and Microbiota-Derived Short-Chain Fatty Acids Promote Bacteriophage Production in the Gut Symbiont Lactobacillus reuteri. Cell Host Microbe 2019, 25, 273–284.e6. [Google Scholar] [CrossRef] [PubMed]
- Caescu, C.I.; Vidal, O.; Krzewinski, F.; Artenie, V.; Bouquelet, S. Bifidobacterium longum requires a fructokinase (Frk; ATP:D-fructose 6-phosphotransferase, EC 2.7.1.4) for fructose catabolism. J. Bacteriol. 2004, 186, 6515–6525. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xia, F.; Wang, J.; Ran, S.; Liang, Y.; Zhou, W.; Huang, Z.; Liang, J. Carbohydrate Metabolism Affects Macrophage-Mediated Killing of Enterococcus faecalis. mSystems 2021, 6, e0043421. [Google Scholar] [CrossRef] [PubMed]
- Isaac, S.; Flor-Duro, A.; Carruana, G.; Puchades-Carrasco, L.; Quirant, A.; Lopez-Nogueroles, M.; Pineda-Lucena, A.; Garcia-Garcera, M.; Ubeda, C. Microbiome-mediated fructose depletion restricts murine gut colonization by vancomycin-resistant Enterococcus. Nat. Commun. 2022, 13, 7718. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Hornung, B.; Renckens, B.; van Hijum, S.A.F.T.; Martins Dos Santos, V.A.P.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. Genomic and functional analysis of. PeerJ 2017, 5, e3698. [Google Scholar] [CrossRef] [PubMed]
- Beisner, J.; Gonzalez-Granda, A.; Basrai, M.; Damms-Machado, A.; Bischoff, S.C. Fructose-Induced Intestinal Microbiota Shift Following Two Types of Short-Term High-Fructose Dietary Phases. Nutrients 2020, 12, 3444. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Alderete, T.L.; Kim, J.S.; Millstein, J.; Gilliland, F.D.; Goran, M.I. High intake of dietary fructose in overweight/obese teenagers associated with depletion of. Gut Microbes 2019, 10, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Goffredo, M.; Mass, K.; Parks, E.J.; Wagner, D.A.; McClure, E.A.; Graf, J.; Savoye, M.; Pierpont, B.; Cline, G.; Santoro, N. Role of Gut Microbiota and Short Chain Fatty Acids in Modulating Energy Harvest and Fat Partitioning in Youth. J. Clin. Endocrinol. Metab. 2016, 101, 4367–4376. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.F.; Butler, R.N.; Brooks, D.A. Intestinal fructose transport and malabsorption in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G202–G206. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Kanmura, S.; Morinaga, Y.; Tanaka, A.; Makino, T.; Fujita, T.; Arima, S.; Sasaki, F.; Nasu, Y.; Tanoue, S.; et al. A high-fructose diet induces epithelial barrier dysfunction and exacerbates the severity of dextran sulfate sodium-induced colitis. Int. J. Mol. Med. 2019, 43, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Packard, C.J.; Boren, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.P.M.M.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef] [PubMed]
- Gligorovska, L.; Teofilovic, A.; Vojnovic Milutinovic, D.; Miladinovic, N.; Kovacevic, S.; Velickovic, N.; Djordjevic, A. Macrophage migration inhibitory factor deficiency aggravates effects of fructose-enriched diet on lipid metabolism in the mouse liver. Biofactors 2021, 47, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Petito, G.; Giacco, A.; Cioffi, F.; Mazzoli, A.; Magnacca, N.; Iossa, S.; Goglia, F.; Senese, R.; Lanni, A. Short-term fructose feeding alters tissue metabolic pathways by modulating microRNAs expression both in young and adult rats. Front. Cell Dev. Biol. 2023, 11, 1101844. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andres-Hernando, A.; Ishimoto, T.; Sanchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, B.; Shen, J.; Bai, M.; Xu, E. Berberine attenuates fructose-induced insulin resistance by stimulating the hepatic LKB1/AMPK/PGC1alpha pathway in mice. Pharm. Biol. 2020, 58, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Lozano, I.; Van der Werf, R.; Bietiger, W.; Seyfritz, E.; Peronet, C.; Pinget, M.; Jeandidier, N.; Maillard, E.; Marchioni, E.; Sigrist, S.; et al. High-fructose and high-fat diet-induced disorders in rats: Impact on diabetes risk, hepatic and vascular complications. Nutr. Metab. 2016, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, N.; Yano, H.; Mizokami, T.; Takahashi, M.; Oyanagi, E.; Suzuki, K. Exercise training attenuates hepatic inflammation, fibrosis and macrophage infiltration during diet induced-obesity in mice. Brain Behav. Immun. 2012, 26, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thone, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, H.; Haass, W.; Castaneda, T.R.; Schurmann, A.; Koebnick, C.; Dombrowski, F.; Otto, B.; Nawrocki, A.R.; Scherer, P.E.; Spranger, J.; et al. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes. Res. 2005, 13, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Sun, S.; Armstrong, M.; Manke, J.; Reisdorph, N.; Adams, V.R.; Kennedy, A.; Zu, Y.; Moustaid-Moussa, N.; Carroll, I.; et al. Beneficial effects of eicosapentaenoic acid on the metabolic profile of obese female mice entails upregulation of HEPEs and increased abundance of enteric Akkermansia muciniphila. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159059. [Google Scholar] [CrossRef] [PubMed]
- Legeza, B.; Marcolongo, P.; Gamberucci, A.; Varga, V.; Banhegyi, G.; Benedetti, A.; Odermatt, A. Fructose, Glucocorticoids and Adipose Tissue: Implications for the Metabolic Syndrome. Nutrients 2017, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Heaney, A.P. Regulation of adipose differentiation by fructose and GluT5. Mol. Endocrinol. 2012, 26, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.C.; Penna-de-Carvalho, A.; Vazquez-Carrera, M.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Short-term administration of GW501516 improves inflammatory state in white adipose tissue and liver damage in high-fructose-fed mice through modulation of the renin-angiotensin system. Endocrine 2015, 50, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Pektas, M.B.; Koca, H.B.; Sadi, G.; Akar, F. Dietary Fructose Activates Insulin Signaling and Inflammation in Adipose Tissue: Modulatory Role of Resveratrol. Biomed. Res. Int. 2016, 2016, 8014252. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Dong, G.; Guo, L.; Graves, D.T. The function of dendritic cells in modulating the host response. Mol. Oral. Microbiol. 2018, 33, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Agrawal, S.; Agrawal, A. High fructose-induced metabolic changes enhance inflammation in human dendritic cells. Clin. Exp. Immunol. 2019, 197, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Kotsiliti, E.; Leone, V.; Schuehle, S.; Govaere, O.; Li, H.; Wolf, M.J.; Horvatic, H.; Bierwirth, S.; Hundertmark, J.; Inverso, D.; et al. Intestinal B cells license metabolic T-cell activation in NASH microbiota/antigen-independently and contribute to fibrosis by IgA-FcR signalling. J. Hepatol. 2023, 79, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Karl, M.; Hasselwander, S.; Zhou, Y.; Reifenberg, G.; Kim, Y.O.; Park, K.S.; Ridder, D.A.; Wang, X.; Seidel, E.; Hovelmeyer, N.; et al. Dual roles of B lymphocytes in mouse models of diet-induced nonalcoholic fatty liver disease. Hepatology 2022, 76, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Bruzzi, S.; Sutti, S.; Giudici, G.; Burlone, M.E.; Ramavath, N.N.; Toscani, A.; Bozzola, C.; Schneider, P.; Morello, E.; Parola, M.; et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic. Biol. Med. 2018, 124, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Ijare, O.B.; Baskin, D.S.; Sharpe, M.A.; Pichumani, K. Metabolism of fructose in B-cells: A (13)C NMR spectroscopy based stable isotope tracer study. Anal. Biochem. 2018, 552, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Ni, D.; Wali, J.A.; Cox, D.A.; Pinget, G.V.; Taitz, J.; Daien, C.I.; Senior, A.; Read, M.N.; Simpson, S.J.; et al. Dietary carbohydrate, particularly glucose, drives B cell lymphopoiesis and function. iScience 2021, 24, 102835. [Google Scholar] [CrossRef] [PubMed]
- Simons, N.; Veeraiah, P.; Simons, P.; Schaper, N.C.; Kooi, M.E.; Schrauwen-Hinderling, V.B.; Feskens, E.J.M.; van der Ploeg, E.; Van den Eynde, M.D.G.; Schalkwijk, C.G.; et al. Effects of fructose restriction on liver steatosis (FRUITLESS); a double-blind randomized controlled trial. Am. J. Clin. Nutr. 2021, 113, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Joh, H.K.; Hur, J.; Song, M.; Zhang, X.; Cao, Y.; Wu, K.; Giovannucci, E.L. Fructose consumption from different food sources and cardiometabolic biomarkers: Cross-sectional associations in US men and women. Am. J. Clin. Nutr. 2023, 117, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ley, S.H.; Sun, Q.; Hu, F.B.; Malik, V.S. Cross-sectional association between sugar-sweetened beverage intake and cardiometabolic biomarkers in US women. Br. J. Nutr. 2018, 119, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Buziau, A.M.; Eussen, S.; Kooi, M.E.; van der Kallen, C.J.H.; van Dongen, M.; Schaper, N.C.; Henry, R.M.A.; Schram, M.T.; Dagnelie, P.C.; van Greevenbroek, M.M.J.; et al. Fructose Intake From Fruit Juice and Sugar-Sweetened Beverages Is Associated with Higher Intrahepatic Lipid Content: The Maastricht Study. Diabetes Care 2022, 45, 1116–1123. [Google Scholar] [CrossRef]
- Parnell, J.A.; Raman, M.; Rioux, K.P.; Reimer, R.A. The potential role of prebiotic fibre for treatment and management of non-alcoholic fatty liver disease and associated obesity and insulin resistance. Liver Int. 2012, 32, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Prinster, A.; Annuzzi, G.; Costagliola, L.; Mangione, A.; Vitelli, A.; Mazzarella, R.; Longobardo, M.; Mancini, M.; Vigorito, C.; et al. Liver fat is reduced by an isoenergetic MUFA diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012, 35, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Cantero, I.; Abete, I.; Monreal, J.I.; Martinez, J.A.; Zulet, M.A. Fruit Fiber Consumption Specifically Improves Liver Health Status in Obese Subjects under Energy Restriction. Nutrients 2017, 9, 667. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Maciejewska, D.; Ryterska, K.; Czerwinka-Rogowska, M.; Jamiol-Milc, D.; Skonieczna-Zydecka, K.; Milkiewicz, P.; Raszeja-Wyszomirska, J.; Stachowska, E. Gut Permeability Might be Improved by Dietary Fiber in Individuals with Nonalcoholic Fatty Liver Disease (NAFLD) Undergoing Weight Reduction. Nutrients 2018, 10, 1793. [Google Scholar] [CrossRef] [PubMed]
- Howlett, J.F.; Betteridge, V.A.; Champ, M.; Craig, S.A.; Meheust, A.; Jones, J.M. The definition of dietary fiber—Discussions at the Ninth Vahouny Fiber Symposium: Building scientific agreement. Food Nutr. Res. 2010, 54, 5750. [Google Scholar] [CrossRef] [PubMed]
- Reiser, S.; Powell, A.S.; Scholfield, D.J.; Panda, P.; Fields, M.; Canary, J.J. Day-long glucose, insulin, and fructose responses of hyperinsulinemic and nonhyperinsulinemic men adapted to diets containing either fructose or high-amylose cornstarch. Am. J. Clin. Nutr. 1989, 50, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- de Melo, F.H.C.; Menezes, F.; de Sousa, J.M.B.; Dos Santos Lima, M.; da Silva Campelo Borges, G.; de Souza, E.L.; Magnani, M. Prebiotic activity of monofloral honeys produced by stingless bees in the semi-arid region of Brazilian Northeastern toward Lactobacillus acidophilus LA-05 and Bifidobacterium lactis BB-12. Food Res. Int. 2020, 128, 108809. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, W.; Zeng, L.Q.; Bai, H.; Li, J.; Zhou, J.; Zhou, G.Y.; Fang, C.W.; Wang, F.; Qin, X.J. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 2020, 36, 101635. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dun, Y.; Zhang, W.; You, B.; Liu, Y.; Fu, S.; Qiu, L.; Cheng, J.; Ripley-Gonzalez, J.W.; Liu, S. Exercise improves lipid droplet metabolism disorder through activation of AMPK-mediated lipophagy in NAFLD. Life Sci. 2021, 273, 119314. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.F.; Csader, S.; Mannisto, V.; Tauriainen, M.M.; Pentikainen, H.; Savonen, K.; Klavus, A.; Koistinen, V.; Hanhineva, K.; Schwab, U. Effects of exercise on NAFLD using non-targeted metabolomics in adipose tissue, plasma, urine, and stool. Sci. Rep. 2022, 12, 6485. [Google Scholar] [CrossRef] [PubMed]
- Ezpeleta, M.; Gabel, K.; Cienfuegos, S.; Kalam, F.; Lin, S.; Pavlou, V.; Song, Z.; Haus, J.M.; Koppe, S.; Alexandria, S.J.; et al. Effect of alternate day fasting combined with aerobic exercise on non-alcoholic fatty liver disease: A randomized controlled trial. Cell Metab. 2023, 35, 56–70.e3. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, A.J.; Fairchild, T.J.; Wang, L.; Keslacy, S.; Kanaley, J.A. Effect of increased physical activity on fructose-induced glycemic response in healthy individuals. Eur. J. Clin. Nutr. 2014, 68, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, H.M.; Ibrahim, M.A.; Amin, E.F.; Abdel-Tawab Ibrahim, S.; Abdel-Wahab, S.; Fouad, Y.M. Allopurinol potentiates the hepatoprotective effect of metformin and vitamin E in fructose-induced fatty liver in rats. Clin. Exp. Hepatol. 2019, 5, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Haleim, E.A.; Bahgat, A.K.; Saleh, S. Resveratrol and fenofibrate ameliorate fructose-induced nonalcoholic steatohepatitis by modulation of genes expression. World J. Gastroenterol. 2016, 22, 2931–2948. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.C.; Bagul, P.K.; Banerjee, S.K. NLRP3 inflammasome drives inflammation in high fructose fed diabetic rat liver: Effect of resveratrol and metformin. Life Sci. 2020, 253, 117727. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz Demirtas, C.; Bircan, F.S.; Pasaoglu, O.T.; Turkozkan, N. The effects of resveratrol on hepatic oxidative stress in metabolic syndrome model induced by high fructose diet. Bratisl. Lek. Listy 2018, 119, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.S.; Li, C.B.; Li, X.Y.; Wu, J.; Li, Y.; Fu, X.; Zhang, Y.; Hu, W.Z. Fisetin Attenuates Metabolic Dysfunction in Mice Challenged with a High-Fructose Diet. J. Agric. Food Chem. 2018, 66, 8291–8298. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; Van Natta, M.L.; Kleiner, D.E.; Clark, J.M.; Kowdley, K.V.; Loomba, R.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; Tonascia, J.; Non-alcoholic Steatohepatitis Clinical Research, N. Vitamin E and changes in serum alanine aminotransferase levels in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2013, 38, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; Izaola, O.; Gomez, S.; Tafur, C.; Gonzalez, G.; Berroa, E.; Mora, N.; Gonzalez, J.M.; de Luis, D.A. Effect of silymarin plus vitamin E in patients with non-alcoholic fatty liver disease. A randomized clinical pilot study. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3118–3124. [Google Scholar] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S.; Polymerou, V.; Katsinelos, P. Effects of combined low-dose spironolactone plus vitamin E vs vitamin E monotherapy on insulin resistance, non-invasive indices of steatosis and fibrosis, and adipokine levels in non-alcoholic fatty liver disease: A randomized controlled trial. Diabetes Obes. Metab. 2017, 19, 1805–1809. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of Vitamin E for Nonalcoholic Steatohepatitis in Patients with Type 2 Diabetes: A Randomized Controlled Trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Cai, Y.; Yuan, Y.; Liu, M.; Zhang, Z.; Xu, Y.; Jiao, P. Efficacy and safety of carnitine supplementation on NAFLD: A systematic review and meta-analysis. Syst. Rev. 2023, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Nakagawa, H.; Enooku, K.; Kudo, Y.; Hayata, Y.; Nakatsuka, T.; Tanaka, Y.; Tateishi, R.; Hikiba, Y.; Misumi, K.; et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut 2018, 67, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Montesano, A.; Senesi, P.; Vacante, F.; Mollica, G.; Benedini, S.; Mariotti, M.; Luzi, L.; Terruzzi, I. L-Carnitine counteracts in vitro fructose-induced hepatic steatosis through targeting oxidative stress markers. J. Endocrinol. Investig. 2020, 43, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arroyo, F.E.; Gonzaga-Sanchez, G.; Silva-Palacios, A.; Roldan, F.J.; Loredo-Mendoza, M.L.; Alvarez-Alvarez, Y.Q.; de Los Santos Coyotl, J.A.; Velez Orozco, K.A.; Tapia, E.; Osorio-Alonso, H.; et al. Osthole Prevents Heart Damage Induced by Diet-Induced Metabolic Syndrome: Role of Fructokinase (KHK). Antioxidants 2023, 12, 1023. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, A.M.; Bentanachs, R.; Sala-Vila, A.; Lazaro, I.; Rodriguez-Morato, J.; Sanchez, R.M.; Laguna, J.C.; Roglans, N.; Alegret, M. KHK, PNPLA3 and PPAR as Novel Targets for the Anti-Steatotic Action of Bempedoic Acid. Biomedicines 2022, 10, 1517. [Google Scholar] [CrossRef] [PubMed]
- Futatsugi, K.; Smith, A.C.; Tu, M.; Raymer, B.; Ahn, K.; Coffey, S.B.; Dowling, M.S.; Fernando, D.P.; Gutierrez, J.A.; Huard, K.; et al. Discovery of PF-06835919: A Potent Inhibitor of Ketohexokinase (KHK) for the Treatment of Metabolic Disorders Driven by the Overconsumption of Fructose. J. Med. Chem. 2020, 63, 13546–13560. [Google Scholar] [CrossRef] [PubMed]
- Kazierad, D.J.; Chidsey, K.; Somayaji, V.R.; Bergman, A.J.; Birnbaum, M.J.; Calle, R.A. Inhibition of ketohexokinase in adults with NAFLD reduces liver fat and inflammatory markers: A randomized phase 2 trial. Med. 2021, 2, 800–813.e3. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [Google Scholar] [CrossRef] [PubMed]
- Simons, N.; Debray, F.G.; Schaper, N.C.; Kooi, M.E.; Feskens, E.J.M.; Hollak, C.E.M.; Lindeboom, L.; Koek, G.H.; Bons, J.A.P.; Lefeber, D.J.; et al. Patients with Aldolase B Deficiency Are Characterized by Increased Intrahepatic Triglyceride Content. J. Clin. Endocrinol. Metab. 2019, 104, 5056–5064. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Liaskou, E.; Fear, J.; Garg, A.; Reynolds, G.; Claridge, L.; Adams, D.H.; Newsome, P.N.; Lalor, P.F. Dysregulated hepatic expression of glucose transporters in chronic disease: Contribution of semicarbazide-sensitive amine oxidase to hepatic glucose uptake. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G1180–G1190. [Google Scholar] [CrossRef] [PubMed]
- Tripp, J.; Essl, C.; Iancu, C.V.; Boles, E.; Choe, J.Y.; Oreb, M. Establishing a yeast-based screening system for discovery of human GLUT5 inhibitors and activators. Sci. Rep. 2017, 7, 6197. [Google Scholar] [CrossRef] [PubMed]
- George Thompson, A.M.; Ursu, O.; Babkin, P.; Iancu, C.V.; Whang, A.; Oprea, T.I.; Choe, J.Y. Discovery of a specific inhibitor of human GLUT5 by virtual screening and in vitro transport evaluation. Sci. Rep. 2016, 6, 24240. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.B.; Chu, X.L.; Jin, Y.X.; Jiang, J.J.; Zhao, X.; Yu, M. Epigallocatechin gallate alleviates high-fat diet-induced hepatic lipotoxicity by targeting mitochondrial ROS-mediated ferroptosis. Front. Pharmacol. 2023, 14, 1148814. [Google Scholar] [CrossRef] [PubMed]
- Satsu, H.; Awara, S.; Unno, T.; Shimizu, M. Suppressive effect of nobiletin and epicatechin gallate on fructose uptake in human intestinal epithelial Caco-2 cells. Biosci. Biotechnol. Biochem. 2018, 82, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Paglicawan, L.; Soomro, S.; Abunofal, O.; Baig, S.; Vanarsa, K.; Hicks, J.; Mohan, C. Epigallocatechin-3-Gallate Dampens Non-Alcoholic Fatty Liver by Modulating Liver Function, Lipid Profile and Macrophage Polarization. Nutrients 2021, 13, 599. [Google Scholar] [CrossRef] [PubMed]
- McMillan, E.A.; Ryu, M.J.; Diep, C.H.; Mendiratta, S.; Clemenceau, J.R.; Vaden, R.M.; Kim, J.H.; Motoyaji, T.; Covington, K.R.; Peyton, M.; et al. Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell 2018, 173, 864–878.e29. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.K.; White, J.R., Jr.; Saulie, B.A. Metformin: A new oral biguanide. Clin. Ther. 1996, 18, 360–371; discussion 359. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Pan, Q.; Xu, Y.; Yang, X. Exenatide improves type 2 diabetes concomitant with non-alcoholic fatty liver disease. Arq. Bras. Endocrinol. Metabol. 2013, 57, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Gao, C.; Bi, Y.; Wu, M.; Li, P.; Shen, S.; Chen, W.; Yin, T.; Zhu, D. Randomized trial comparing the effects of gliclazide, liraglutide, and metformin on diabetes with non-alcoholic fatty liver disease. J. Diabetes 2017, 9, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Cheng, K.; Xu, S.; Li, S.; Zhou, Y.; Zhou, S.; Kong, R.; Li, L.; Li, J.; Feng, J.; et al. Metformin and Diammonium Glycyrrhizinate Enteric-Coated Capsule versus Metformin Alone versus Diammonium Glycyrrhizinate Enteric-Coated Capsule Alone in Patients with Nonalcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus. Gastroenterol. Res. Pract. 2017, 2017, 8491742. [Google Scholar] [CrossRef] [PubMed]
- Yabiku, K.; Mutoh, A.; Miyagi, K.; Takasu, N. Effects of Oral Antidiabetic Drugs on Changes in the Liver-to-Spleen Ratio on Computed Tomography and Inflammatory Biomarkers in Patients with Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Clin. Ther. 2017, 39, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Zheng, Z.; Zhang, D.; He, S.; Shen, J. Efficacy of liraglutide in treating type 2 diabetes mellitus complicated with non-alcoholic fatty liver disease. Biosci. Rep. 2018, 38, BSR20181304. [Google Scholar] [CrossRef] [PubMed]
- Zsori, G.; Illes, D.; Ivany, E.; Kosar, K.; Holzinger, G.; Tajti, M.; Palinkas, E.; Szabovik, G.; Nagy, A.; Palko, A.; et al. In New-Onset Diabetes Mellitus, Metformin Reduces Fat Accumulation in the Liver, But Not in the Pancreas or Pericardium. Metab. Syndr. Relat. Disord. 2019, 17, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Metformin protects against the development of fructose-induced steatosis in mice: Role of the intestinal barrier function. Lab. Investig. 2012, 92, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jia, Z.; Li, Y.R.; Danelisen, I. Molecular mechanisms of action of metformin: Latest advances and therapeutic implications. Clin. Exp. Med. 2023, 23, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Karise, I.; Ornellas, F.; Barbosa-da-Silva, S.; Matsuura, C.; Del Sol, M.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Liver and Metformin: Lessons of a fructose diet in mice. Biochim. Open 2017, 4, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Nassif, R.M.; Chalhoub, E.; Chedid, P.; Hurtado-Nedelec, M.; Raya, E.; Dang, P.M.; Marie, J.C.; El-Benna, J. Metformin Inhibits ROS Production by Human M2 Macrophages via the Activation of AMPK. Biomedicines 2022, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol. Cell Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Qing, L.; Fu, J.; Wu, P.; Zhou, Z.; Yu, F.; Tang, J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am. J. Transl. Res. 2019, 11, 655–668. [Google Scholar] [PubMed]
- Meloni, A.R.; DeYoung, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic beta-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol 2021, 12, 721135. [Google Scholar] [CrossRef] [PubMed]
- Galderisi, A.; Giannini, C.; Van Name, M.; Caprio, S. Fructose Consumption Contributes to Hyperinsulinemia in Adolescents with Obesity Through a GLP-1-Mediated Mechanism. J. Clin. Endocrinol. Metab. 2019, 104, 3481–3490. [Google Scholar] [CrossRef] [PubMed]
- Yunker, A.G.; Luo, S.; Jones, S.; Dorton, H.M.; Alves, J.M.; Angelo, B.; DeFendis, A.; Pickering, T.A.; Monterosso, J.R.; Page, K.A. Appetite-Regulating Hormones Are Reduced After Oral Sucrose vs Glucose: Influence of Obesity, Insulin Resistance, and Sex. J. Clin. Endocrinol. Metab. 2021, 106, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Mita, T.; Azuma, K.; Ebato, C.; Goto, H.; Nomiyama, T.; Fujitani, Y.; Hirose, T.; Kawamori, R.; Watada, H. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 2010, 59, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Gao, H.; Guo, Y.X.; Wang, B.Y.; Hua, R.X.; Gao, L.; Shang, H.W.; Lu, X.; Xu, J.D. GLP-1 and Underlying Beneficial Actions in Alzheimer’s Disease, Hypertension, and NASH. Front. Endocrinol. 2021, 12, 721198. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.A.; Brown, J.D.; Ayala, J.E.; Stoffers, D.A.; Sandoval, D.A.; Seeley, R.J.; Ayala, J.E. The glucagon-like peptide-1 receptor in the ventromedial hypothalamus reduces short-term food intake in male mice by regulating nutrient sensor activity. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E651–E662. [Google Scholar] [CrossRef] [PubMed]
- van Bloemendaal, L.; RG, I.J.; Ten Kulve, J.S.; Barkhof, F.; Konrad, R.J.; Drent, M.L.; Veltman, D.J.; Diamant, M. GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes 2014, 63, 4186–4196. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.A.; Ayala, J.; Drucker, D.J.; Ayala, J.E. Central glucagon-like peptide 1 receptor-induced anorexia requires glucose metabolism-mediated suppression of AMPK and is impaired by central fructose. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E677–E685. [Google Scholar] [CrossRef] [PubMed]
- Ghidewon, M.; Wald, H.S.; McKnight, A.D.; De Jonghe, B.C.; Breen, D.M.; Alhadeff, A.L.; Borner, T.; Grill, H.J. Growth differentiation factor 15 (GDF15) and semaglutide inhibit food intake and body weight through largely distinct, additive mechanisms. Diabetes Obes. Metab. 2022, 24, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, K.; Kowalczyk, K.; Cwynar, M.; Czapla, D.; Czarkowski, W.; Kmita, D.; Nowak, A.; Madej, P. The Role of Glp-1 Receptor Agonists in Insulin Resistance with Concomitant Obesity Treatment in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2022, 23, 4334. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Song, G.Y.; Ren, L.P.; Ma, H.J.; Ma, B.Q.; Chen, S.C. beta-catenin mediates the effect of GLP-1 receptor agonist on ameliorating hepatic steatosis induced by high fructose diet. Eur. J. Histochem. 2020, 64, 3160. [Google Scholar] [CrossRef] [PubMed]
- Li, S.L.; Wang, Z.M.; Xu, C.; Che, F.H.; Hu, X.F.; Cao, R.; Xie, Y.N.; Qiu, Y.; Shi, H.B.; Liu, B.; et al. Liraglutide Attenuates Hepatic Ischemia-Reperfusion Injury by Modulating Macrophage Polarization. Front. Immunol. 2022, 13, 869050. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Laker, R.C.; Mather, K.; Nawrocki, A.; Oldham, S.; Boland, B.B.; Lewis, H.; Conway, J.; Naylor, J.; Guionaud, S.; et al. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat. Metab. 2020, 2, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, S.; Ospina, E.; Shabandri, O.; Lank, D.; Akakpo, J.Y.; Zhao, Z.; Yang, M.; Wu, J.; Jaeschke, H.; et al. Fructose Protects Against Acetaminophen-Induced Hepatotoxicity Mainly by Activating the Carbohydrate-Response Element-Binding Protein alpha-Fibroblast Growth Factor 21 Axis in Mice. Hepatol. Commun. 2021, 5, 992–1008. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, N.; Sandboge, S.; Kaartinen, N.E.; Mannisto, S.; Eriksson, J.G. Higher fructose intake is inversely associated with risk of nonalcoholic fatty liver disease in older Finnish adults. Am. J. Clin. Nutr. 2014, 100, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, V.Z.; Dall’Alba, V. Fructose intake is not associated to the risk of hepatic fibrosis in patients with Non-Alcoholic Fatty Liver Disease (NAFLD). Clin. Nutr. 2021, 40, 4275–4283. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Li, L.; Ecelbarger, C.M. Sex differences in renal and metabolic responses to a high-fructose diet in mice. Am. J. Physiol. Renal Physiol. 2015, 308, F400–F410. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Jacot-Descombes, D.; Lecoultre, V.; Fielding, B.A.; Carrel, G.; Le, K.A.; Schneiter, P.; Bortolotti, M.; Frayn, K.N.; Tappy, L. Sex differences in lipid and glucose kinetics after ingestion of an acute oral fructose load. Br. J. Nutr. 2010, 104, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Pektas, M.B.; Sadi, G.; Akar, F. Long-Term Dietary Fructose Causes Gender-Different Metabolic and Vascular Dysfunction in Rats: Modulatory Effects of Resveratrol. Cell Physiol. Biochem. 2015, 37, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Hyer, M.M.; Dyer, S.K.; Kloster, A.; Adrees, A.; Taetzsch, T.; Feaster, J.; Valdez, G.; Neigh, G.N. Sex modifies the consequences of extended fructose consumption on liver health, motor function, and physiological damage in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R903–R911. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Hu, W.; Ma, T.; Hu, M.; Tong, X.; Zhang, F.; Shi, J.; Xu, X.; Li, X.; Shao, L.R.; et al. Long-term androgen excess induces insulin resistance and non-alcoholic fatty liver disease in PCOS-like rats. J. Steroid Biochem. Mol. Biol. 2021, 208, 105829. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Kato, M.; Yamasaki, A.; Kuwano, A.; Suzuki, H.; Kohjima, M.; Ogawa, Y. Effects of high fructose intake on liver injury progression in high fat diet induced fatty liver disease in ovariectomized female mice. Food Chem. Toxicol. 2018, 118, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Hsia, S.M.; Chiang, Y.F.; Chen, H.Y.; Ali, M.; Wang, P.S.; Wang, K.L. Effect of High-Fructose Diet-Induced Metabolic Syndrome on the Pituitary-Gonadal Axis in Male Rats. Biomedicines 2022, 10, 3009. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodge, M.; Dykes, R.; Kennedy, A. Regulation of Fructose Metabolism in Nonalcoholic Fatty Liver Disease. Biomolecules 2024, 14, 845. https://doi.org/10.3390/biom14070845
Lodge M, Dykes R, Kennedy A. Regulation of Fructose Metabolism in Nonalcoholic Fatty Liver Disease. Biomolecules. 2024; 14(7):845. https://doi.org/10.3390/biom14070845
Chicago/Turabian StyleLodge, Mareca, Rachel Dykes, and Arion Kennedy. 2024. "Regulation of Fructose Metabolism in Nonalcoholic Fatty Liver Disease" Biomolecules 14, no. 7: 845. https://doi.org/10.3390/biom14070845