
Calcium and Non-Penetrating Traumatic Brain Injury: A Proposal for the Implementation of an Early Therapeutic Treatment for Initial Head Insults


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
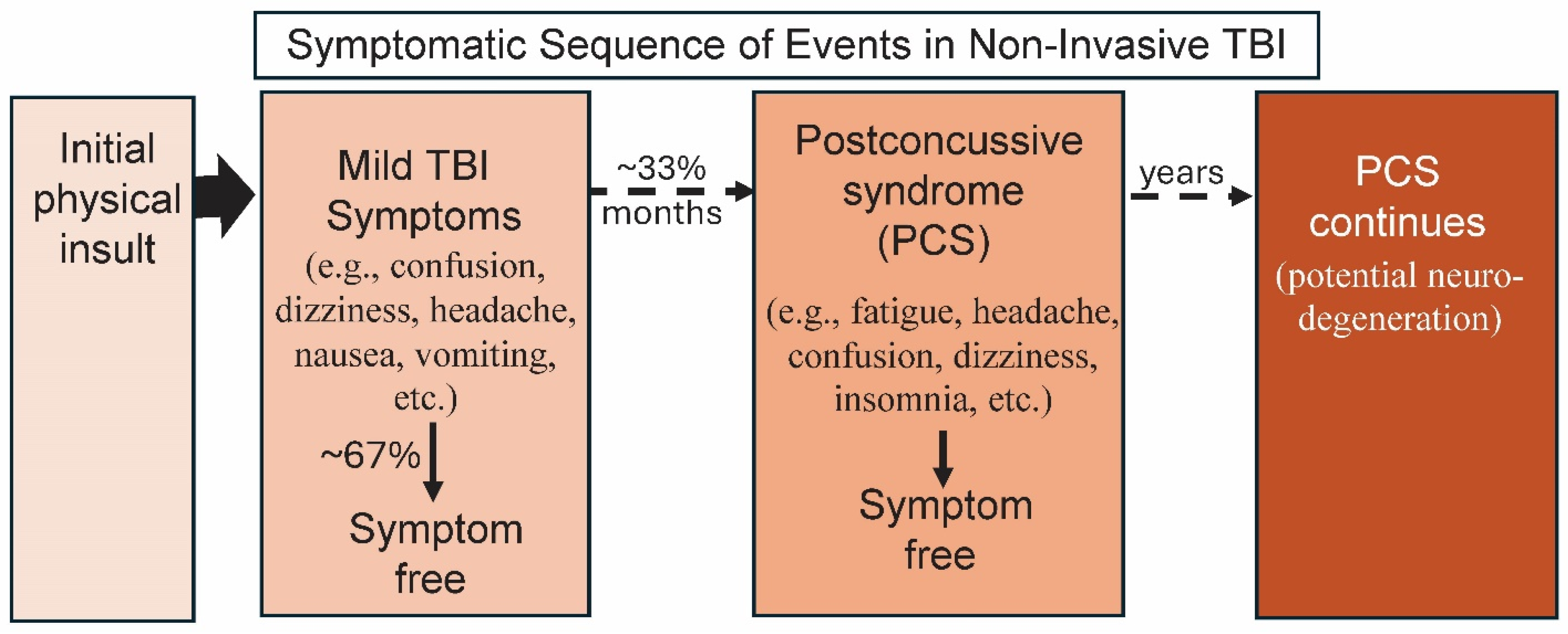
:1. TBI: Its Initiation and Long-Term Effects
2. Current Treatments for mTBI
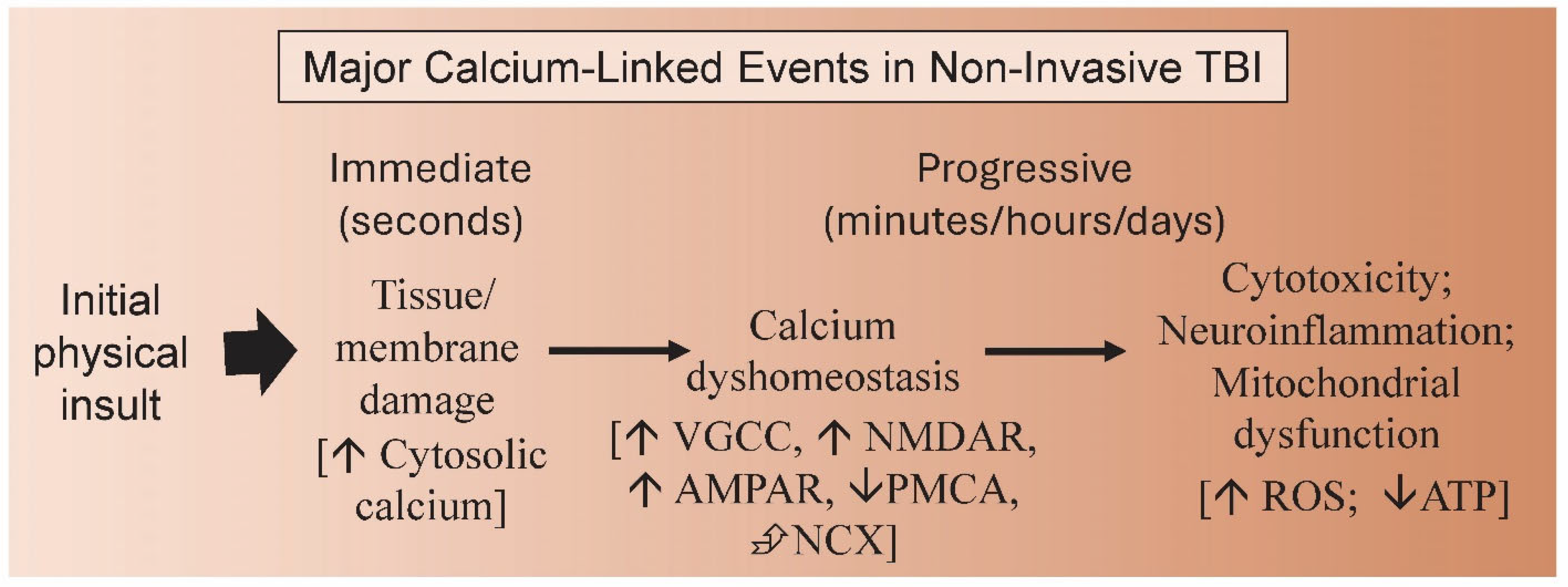
3. Calcium Dysregulation and TBI
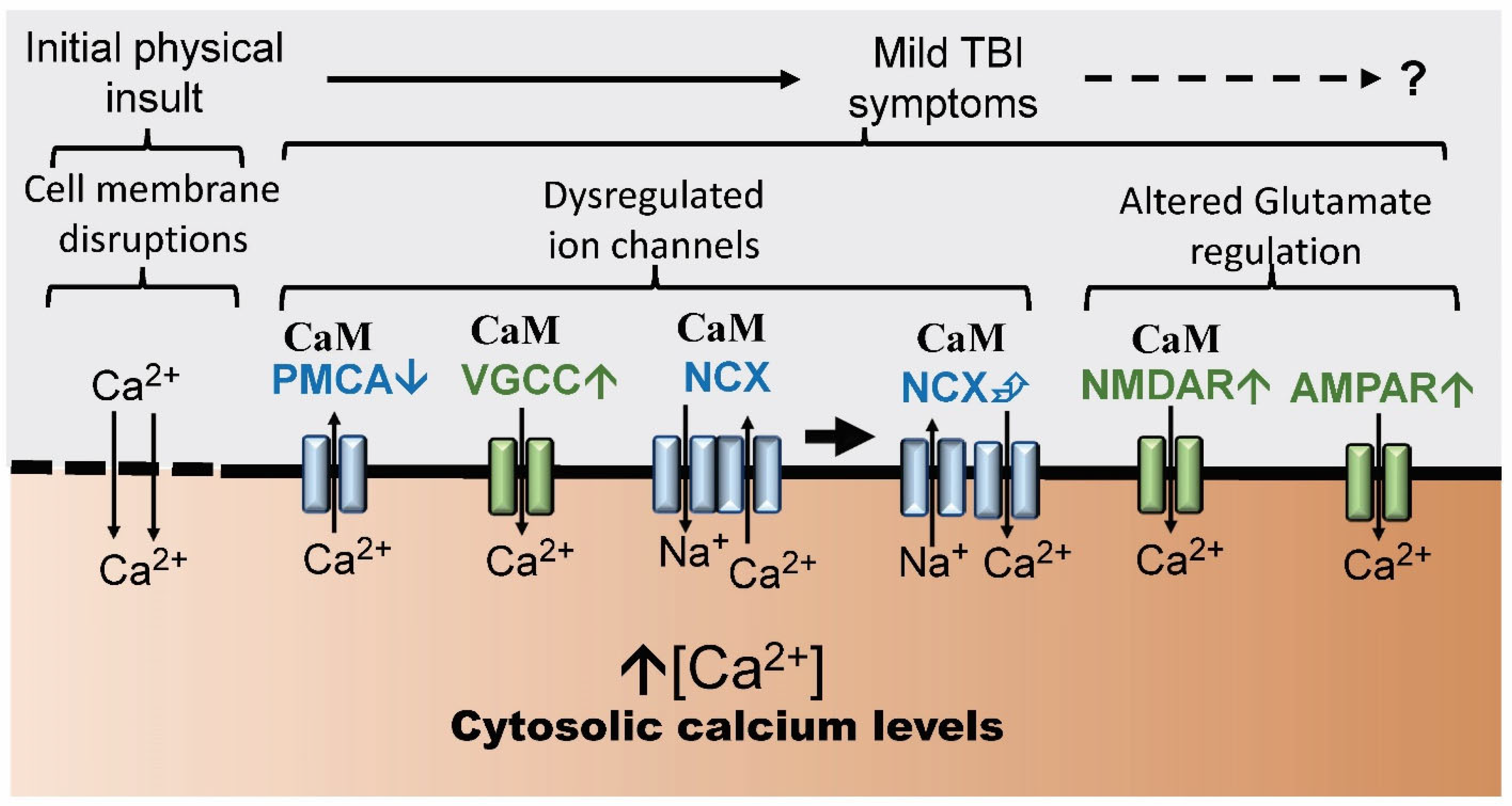
4. Calcium Ion Channels and TBI
5. The Downstream Role of Calcium Signaling: Calcium-Binding Proteins
6. Calmodulin-Regulated Ion Channels and TBI
7. CaMKII, Calcineurin and TBI
8. Discussion
9. mTBI: A Therapeutic Proposal
10. Some Potential Treatments
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petersen, A.; Soderstrom, M.; Saha, B.; Sharma, P. Animal models of traumatic brain injury: A review of pathophysiology to biomarkers and treatments. Exp. Brain Res. 2021, 239, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- G.B.D. Traumatic Brain Injury and Spinal Cord Injury Collaborators. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 56–87. [Google Scholar] [CrossRef] [PubMed]
- Izzy, S.; Chen, P.M.; Tahir, Z.; Grashow, R.; Radmanesh, F.; Cote, D.J.; Yahya, T.; Dhand, A.; Taylor, H.; Shih, S.L.; et al. Association of Traumatic Brain Injury with the Risk of Developing Chronic Cardiovascular, Endocrine, Neurological, and Psychiatric Disorders. JAMA Netw. Open 2022, 5, e229478. [Google Scholar] [CrossRef] [PubMed]
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology associated with traumatic brain injury: Current treatments and potential novel therapeutics. Cell Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef]
- Freire, M.A.M.; Rocha, G.S.; Bittencourt, L.O.; Falcao, D.; Lima, R.R.; Cavalcanti, J.R.L.P. Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far? Biology 2023, 12, 1139. [Google Scholar] [CrossRef] [PubMed]
- Ghaith, H.S.; Nawar, A.A.; Gabra, M.D.; Abdelrahman, M.E.; Nafady, M.H.; Bahbah, E.I.; Ebada, M.A.; Ashraf, G.M.; Negida, A.; Barreto, G.E. A Literature Review of Traumatic Brain Injury Biomarkers. Mol. Neurobiol. 2022, 59, 4141–4158. [Google Scholar] [CrossRef]
- Lynch, D.G.; Narayan, R.K.; Li, C. Multi-Mechanistic Approaches to the Treatment of Traumatic Brain Injury: A Review. J. Clin. Med. 2023, 12, 2179. [Google Scholar] [CrossRef]
- Martinez, B.I.; Mousa, G.A.; Fleck, K.; Macculloch, T.; Stabenfeldt, S.E. Uncovering temporospatial sensitive TBI targeting strategies via in vivo phage display. Sci. Adv. 2022, 8, eabo5047. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef]
- Deshetty, U.M.; Periyasamy, P. Potential Biomarkers in Experimental Animal Models for Traumatic Brain Injury. J. Clin. Med. 2023, 12, 3923. [Google Scholar] [CrossRef]
- Levin, H.S.; Diaz-Arrastia, R.R. Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol. 2015, 14, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; McKee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Prim. 2016, 2, 16084. [Google Scholar] [CrossRef] [PubMed]
- Kristman, V.L.; Borg, J.; Godbolt, A.K.; Salmi, L.R.; Cancelliere, C.; Carroll, L.J.; Holm, L.W.; Nygren-de Boussard, C.; Hartvigsen, J.; Abara, U.; et al. Methodological issues and research recommendations for prognosis after mild traumatic brain injury: Results of the international collaboration on mild traumatic brain injury prognosis. Arch. Phys. Med. Rehabil. 2014, 95, S265–S277. [Google Scholar] [CrossRef] [PubMed]
- Bey, T.; Ostick, B. Second impact syndrome. West. J. Emerg. Med. 2009, 10, 6–10. [Google Scholar] [PubMed]
- Bazarian, J.J.; Wong, T.; Harris, M.; Leahey, N.; Mookerjee, S.; Dombovy, M. Epidemiology and predictors of post-concussive syndrome after minor head injury in an emergency population. Brain Inj. 1999, 13, 173–189. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic Brain Injury: Oxidative Stress and Neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Radabaugh, H.L.; Ferguson, A.R.; Bramlett, H.M.; Dietrich, W.D. Increasing Rigor of Preclinical Research to Maximize Opportunities for Translation. Neurotherapeutics 2023, 6, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Ntikas, M.; Stewart, W.; Ietswaart, M.; Hunter, A.M.; Maas, A.I.R.; Menon, D.K.; Wilson, L.; CENTER-TBI participants and investigators. Contrasting Characteristics and Outcomes of Sports-Related and Non-Sports-Related Traumatic Brain Injury. JAMA Network Open 2024, 7, e2353318. [Google Scholar] [CrossRef] [PubMed]
- McDaid, J.; Briggs, C.A.; Barrington, N.M.; Peterson, D.A.; Kozlowski, D.A.; Stutzmann, G.E. Sustained Hippocampal Synaptic Pathophysiology Following Single and Repeated Closed-Head Concussive Impacts. Front. Cell. Neurosci. 2021, 15, 652721. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Kochanek, P.M. Pharmacotherapy for Traumatic Brain Injury: The Next Generation of Clinical Trials. Neurotherapeutics 2023, 20, 1428–1432. [Google Scholar] [CrossRef]
- Syzdykbayev, M.; Kazymov, M.; Aubakirov, M.; Kurmangazina, A.; Kairkhanov, E.; Kazangapov, R.; Bryzhakhina, Z.; Imangazinova, S.; Sheinin, A. A Modern Approach to the Treatment of Traumatic Brain Injury. Medicines 2024, 11, 10. [Google Scholar] [CrossRef]
- Yan, A.; Torpey, A.; Morrisroe, E.; Andraous, W.; Costa, A.; Bergese, S. Clinical Management in Traumatic Brain Injury. Biomedicines 2024, 12, 781. [Google Scholar] [CrossRef] [PubMed]
- Mayo Clinic Website. 2024. Available online: https://www.mayoclinic.org/diseases-conditions/traumatic-brain-injury/diagnosis-treatment/drc-20378561 (accessed on 23 May 2024).
- Young, W. Role of calcium in central nervous system injuries. J. Neurotrauma 1992, 9, S9–S25. [Google Scholar]
- Perry, D.C.; Sturm, V.E.; Peterson, M.J.; Pieper, C.F.; Bullock, T.; Boeve, B.F.; Miller, B.L.; Guskiewicz, K.M.; Berger, M.S.; Kramer, J.H.; et al. Association of traumatic brain injury with subsequent neurological and psychiatric disease: A meta-analysis. J. Neurosurg. 2016, 124, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.A.; Deshpande, L.S.; Sombati, S.; Baranova, A.; Wilson, M.S.; Hamm, R.J.; DeLorenzo, R.J. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur. J. Neurosci. 2008, 27, 1659–1672. [Google Scholar] [CrossRef]
- Lima, R.R.; Oliveira, A.C.A.; Fernandes, R.M.; Nascimento, P.C.; Freire, M.A.M.; Gomes-Leal, W. Inflammatory Response and Secondary White Matter Damage to the Corpus Callosum after Focal Striatal Stroke in Rats. Int. J. Mol. Sci. 2022, 23, 3179. [Google Scholar] [CrossRef]
- Freire, M.A.M.; Lima, R.R.; Bittencourt, L.O.; Guimaraes, J.S.; Falcao, D.; Gomes-Leal, W. Astrocytosis, Inflammation, Axonal Damage and Myelin Impairment in the Internal Capsule following Striatal Ischemic Injury. Cells 2023, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Hoffe, B.; Holahan, M.R. Hyperacute Excitotoxic Mechanisms and Synaptic Dysfunction Involved in Traumatic Brain Injury. Front. Mol. Neurosci. 2022, 15, 831825. [Google Scholar] [CrossRef]
- O’Day, D.H. The Complex Interplay between Toxic Hallmark Proteins, Calmodulin-Binding Proteins, Ion Channels, and Receptors Involved in Calcium Dyshomeostasis in Neurodegeneration. Biomolecules 2024, 14, 173. [Google Scholar] [CrossRef]
- O’Day, D.H. Calmodulin binding domains in critical risk proteins involved in neurodegeneration. Curr. Issues Mol. Biol. 2022, 44, 5802–5814. [Google Scholar] [CrossRef]
- Fineman, I.; Hovda, D.A.; Smith, M.; Yoshino, A.; Becker, D.P. Concussive brain injury is associated with a prolonged accumulation of calcium: A 45Ca autoradiographic study. Brain Res. 1993, 624, 94–102. [Google Scholar] [CrossRef]
- Pettus, E.H.; Christman, C.W.; Giebel, M.L.; Povlishock, J.T. Traumatically induced altered membrane permeability: Its relationship to traumatically induced reactive axonal change. J. Neurotrauma 1994, 11, 507–522. [Google Scholar] [CrossRef]
- Wolf, J.A.; Stys, P.K.; Lusardi, T.; Meaney, D.; Smith, D.H. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 2001, 21, 1923–1930. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef]
- Song, J.L.; Westover, M.B.; Zhang, R. A mechanistic model of calcium homeostasis leading to occurrence and propagation of secondary brain injury. J. Neurophysiol. 2022, 128, 1168–1180. [Google Scholar] [CrossRef]
- Weber, J.T. Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 2012, 3, 60. [Google Scholar] [CrossRef]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef]
- Hayes, R.L.; Jenkins, L.W.; Lyeth, B.G. Neurotransmitter-mediated mechanisms of traumatic brain injury: Acetylcholine and excitatory amino acids. J. Neurotrauma 1992, 9, S173–S187. [Google Scholar]
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. 2010, 113, 564–570. [Google Scholar] [CrossRef]
- Park, E.; Bell, J.D.; Baker, A.J. Traumatic brain injury: Can the consequences be stopped? CMAJ 2008, 178, 1163–1170. [Google Scholar] [CrossRef]
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef]
- Langham, J.; Goldfrad, C.; Teasdale, G.; Shaw, D.; Rowan, K.; Group, C.I. Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst. Rev. 2003, 2010, CD000565. [Google Scholar] [CrossRef]
- Kostron, H.; Twerdy, K.; Stampfl, G.; Mohsenipour, I.; Fischer, J.; Grunert, V. Treatment of the traumatic cerebral vasospasm with the calcium channel blocker nimodipine: A preliminary report. Neurol. Res. 2016, 6, 29–32. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Emerging treatments for traumatic brain injury. Expert Opin. Emerg. Drugs 2009, 14, 67–84. [Google Scholar] [CrossRef]
- Lee, L.L.; Galo, E.; Lyeth, B.G.; Muizelaar, J.P.; Berman, R.F. Neuroprotection in the rat lateral fluid percussion model of traumatic brain injury by SNX-185, an N-type voltage-gated calcium channel blocker. Exp. Neurol. 2004, 190, 70–78. [Google Scholar] [CrossRef]
- Gurkoff, G.; Shahlaie, K.; Lyeth, B.; Berman, R. Voltage-gated calcium channel antagonists and traumatic brain injury. Pharmaceuticals 2013, 6, 788–812. [Google Scholar] [CrossRef]
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef]
- Monroe, D.C.; Thomas, E.A.; Cecchi, N.J.; Granger, D.A.; Hicks, J.W.; Small, S.L. Salivary S100 calcium-binding protein beta (S100B) and neurofilament light (NfL) after acute exposure to repeated head impacts in collegiate water polo players. Sci. Rep. 2022, 12, 3439. [Google Scholar] [CrossRef]
- Pineda, J.A.; Wang, K.K.W.; Hayes, R.L. Biomarkers of proteolytic damage following traumatic brain injury. Brain Pathol. 2004, 14, 202–209. [Google Scholar] [CrossRef]
- Zhang, Z.; Ottens, A.K.; Golden, E.C.; Hayes, R.L.; Wang, K.K.W. Using calmodulin-affinity capture to study the rat brain calmodulin binding proteome and its vulnerability to calpain and caspase proteolysis. Calcium Bind. Proteins 2004, 2, 125–134. [Google Scholar]
- Zhang, Z.; Kobeissy, F.H.; Ottens, A.K.; Martínez, J.A.; Wang, K.K. Calmodulin-binding proteome in the brain. Methods Mol. Biol. 2009, 566, 181–190. [Google Scholar]
- Toescu, E.C. Apoptosis and cell death in neuronal cells: Where does Ca2+ fit in? Cell Calcium 1998, 24, 387–403. [Google Scholar] [CrossRef]
- Saatman, K.E.; Creed, J.; Raghupathi, R. Calpain as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics 2010, 7, 31–42. [Google Scholar] [CrossRef]
- Baudry, M.; Luo, Y.L.; Bi, X. Calpain-2 Inhibitors as Therapy for Traumatic Brain Injury. Neurotherapeutics 2023, 20, 1592–1602. [Google Scholar] [CrossRef]
- Means, A.; Dedman, J. Calmodulin—An intracellular calcium receptor. Nature 1980, 285, 73–77. [Google Scholar] [CrossRef]
- Klee, C.B.; Crouch, T.H.; Richman, P.G. Calmodulin. Annu. Rev. Biochem. 1980, 49, 489–515. [Google Scholar] [CrossRef]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [Google Scholar] [CrossRef]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Westerlund, A.M.; Delemotte, L. Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput. Biol. 2018, 14, e1006072. [Google Scholar] [CrossRef]
- Kirberger, M.; Yang, J.J. Structural aspects and prediction of calmodulin-binding proteins. Int. J. Mol. Sci. 2021, 22, 308. [Google Scholar]
- Grant, B.M.M.; Enomoto, M.; Ikura, M.; Marshall, C.B. A non-canonical calmodulin target motif comprising a polybasic region and lapidated terminal residue regulates localization. Int. J. Mol. Sci. 2020, 21, 2751. [Google Scholar] [CrossRef]
- Jurado, L.A.; Chockalingam, P.S.; Jarrett, H.W. Apocalmodulin. Physiol Rev. 1999, 3, 661–682. [Google Scholar] [CrossRef]
- Boczek, T.; Sobolczyk, M.; Mackiewicz, J.; Lisek, M.; Ferenc, B.; Guo, F.; Zylinska, L. Crosstalk among calcium ATPases: PMCA, SERCA and SPCA in mental diseases. Int. J. Mol. Sci. 2021, 22, 2785. [Google Scholar] [CrossRef]
- Nitsche, J.; Josts, I.; Heidemann, J.; Mertens, H.D.; Maric, S.; Moulin, M.; Haertlein, M.; Busch, S.; Forsyth, V.T.; Tidow, H.; et al. Structural basis for activation of plasma-membrane Ca2+-ATPase by calmodulin. Commun. Biol. 2018, 1, 206. [Google Scholar] [CrossRef]
- Krebs, J. Structure, function and regulation of the plasma membrane calcium pump in health and disease. Int. J. Mol. Sci. 2022, 23, 1027. [Google Scholar] [CrossRef]
- Luo, P.; Li, X.; Wu, X.; Dai, S.; Yang, Y.; Xu, H.; Jing, D.; Rao, W.; Xu, H.; Lu, H.; et al. Preso regulates NMDA receptor-mediated excitotoxicity via modulating nitric oxide and calcium responses after traumatic brain injury. Cell Death. Dis. 2019, 10, 496. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Cerpa, W. Regulation of Phosphorylated State of NMDA Receptor by STEP61 Phosphatase after Mild-Traumatic Brain Injury: Role of Oxidative Stress. Antioxidants 2021, 10, 1575. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G.J.; Popescu, G.K. Calcium- and calmodulin-dependent inhibition of NMDA receptor currents. Biophys. J. 2024, 123, 277–293. [Google Scholar] [CrossRef]
- Ehlers, M.D.; Zhang, S.; Bernhardt, J.P.; Huganir, R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 1996, 84, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Vogel, H.J. Structural basis for the regulation of L-type voltage-gated calcium channels: Interactions between the N-terminal cytoplasmic domain and Ca2+-calmodulin. Front. Mol. Neurosci. 2012, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G.J.; Popescu, G.K. Resident Calmodulin Primes NMDA Receptors for Ca2+-Dependent Inactivation. Biophys. J. 2017, 113, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Vieira, D.C.O.; Yue, D.T.; Ben-Johny, M.; Dick, I.E. The molecular basis of the inhibition of CaV1 calcium-dependent inactivation by the distal carboxy tail. J. Biol. Chem. 2021, 296, 100502. [Google Scholar] [CrossRef] [PubMed]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef]
- Antunes, F.T.T.; De Souza, A.H.; Figueira, J.; Binda, N.S.; Carvalho, V.P.R.; Vieira, L.B.; Gomez, M.V. Targeting N-type calcium channels in young-onset of some neurological diseases. Front. Cell Dev. Biol. 2022, 10, 1090765. [Google Scholar] [CrossRef]
- Taiakina, V.; Boone, A.N.; Fux, J.; Senatore, A.; Weber-Adrian, D.; Guillemette, J.G.; Spafford, J.D. The calmodulin-binding, short linear motif, NSCaTE is conserved in L-type channel ancestors of vertebrate Cav1.2 and Cav1.3 channels. PLoS ONE 2013, 8, e61765. [Google Scholar] [CrossRef] [PubMed]
- Yaduvanshi, S.; Ero, R.; Kumar, V. The mechanism of complex formation between calmodulin and voltage gated calcium channels revealed by molecular dynamics. PLoS ONE 2021, 16, e0258112. [Google Scholar] [CrossRef]
- Chou, A.C.; Ju, Y.T.; Pan, C.Y. Calmodulin Interacts with the Sodium/Calcium Exchanger NCX1 to Regulate Activity. PLoS ONE 2015, 10, e0138856. [Google Scholar] [CrossRef]
- Thibodeau, S.; Yang, W.; Sharma, S.; Lytton, J. Calmodulin binds and modulates K+-dependent Na+/Ca2+-exchanger isoform 4, NCKX4. J Biol Chem. 2021, 296, 100092. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Gu, Z.; Wang, D.; Wang, T.; Wang, Z.; Tao, L. Increased expression of calcium/calmodulin-dependent protein kinase typeII subunit delta after rat traumatic brain injury. J. Mol. Neurosci. 2012, 46, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, A.G.; Gunasekaran, S.; Jacob, R.S.; Omkumar, R.V. Role of Ca2C/Calmodulin-Dependent Protein Kinase Type II in Mediating Function and Dysfunction at Glutamatergic Synapses. Front. Mol. Neurosci. 2022, 15, 855752. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimers Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Bales, J.W.; Ma, X.; Yan, H.Q.; Jenkins, L.W.; Dixon, C.E. Expression of protein phosphatase 2B (calcineurin) subunit A isoforms in rat hippocampus after traumatic brain injury. J. Neurotrauma. 2010, 27, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Holstein, D.M.; Aime, S.; Bollo, M.; Lechleiter, J.D. Calcineurin β protects brain after injury by activating the unfolded protein response. Neurobiol. Dis. 2016, 94, 139–156. [Google Scholar] [CrossRef]
- Kim, S.; Ziff, E.B. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 2014, 12, e1001900. [Google Scholar] [CrossRef] [PubMed]
- Reese, L.C.; Taglialatela, G. A role for calcineurin in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L.; et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010, 30, 2636–2649. [Google Scholar] [CrossRef]
- Rozkalne, A.; Hyman, B.T.; Spires-Jones, T.L. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 2011, 41, 650–654. [Google Scholar] [CrossRef]
- Hossain, I.; Mohammadian, M.; Maanpää, H.R.; Takala, R.S.; Tenovuo, O.; van Gils, M.; Hutchinson, P.; Menon, D.K.; Newcombe, V.F.; Posti, J.P.; et al. Plasma neurofilament light admission levels and development of axonal pathology in mild traumatic brain injury. BMC Neurol. 2023, 23, 304. [Google Scholar] [CrossRef] [PubMed]
- Sahler, C.S.; Greenwald, B.D. Traumatic brain injury in sports: A review. Rehabil. Res. Pract. 2012, 2012, 659652. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Mahon, S.; Hume, P.; Starkey, N.; Barker-Collo, S.; Jones, K.; Majdan, M.; Feigin, V.L. Incidence of Sports-Related Traumatic Brain Injury of All Severities: A Systematic Review. Neuroepidemiology 2020, 54, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Lanzetti, S.; Di Biase, V. Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives. Molecules 2022, 27, 1312. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Gao, S.; Yan, N. Structural biology of voltage-gated calcium channels. Channels 2024, 18, 2290807. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.P.; Cao, L.L.; Yang, Y.; Wang, P. Calcium ions aggravate Alzheimer’s disease through the aberrant activation of neuronal networks, leading to synaptic and cognitive deficits. Front. Mol. Neurosci. 2021, 12, 4634. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.H.; Kim, J.H.; Shim, J.S.; Kwon, H.J. A novel Ca2+/calmodulin antagonist HBC inhibits angiogenesis and down-regulated hypoxia-inducible factor. J. Biol. Chem. 2010, 285, 25867–25874. [Google Scholar] [CrossRef]
- Yuan, K.; Yong, S.; Xu, F.; Zhou, T.; McDonald, J.M.; Chen, Y. Calmodulin antagonists promote TRA-8 therapy of resistant pancreatic cancer. Oncotarget 2015, 6, 25308–25319. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yin, Y.X.; Mahmood, Q.; Wang, X.J.; Gao, Y.P.; Gou, G.J.; Ahmed, M.M.; Kohji, F.; Du, Y.Z.; Han, F. Calmodulin inhibitor ameliorates cognitive dysfunction via inhibiting nitrosative stress and NLRP3 signaling in mice with bilateral carotid artery stenosis. CNS Neurosci. Ther. 2017, 23, 818–826. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2024. Alzheimers Dement. 2024, 10, e12465. [Google Scholar] [CrossRef]
- Taglialatella, G.; Rastellini, C.; Cicalese, L. Reduced incidence of dementia in solid organ transplant patients treated with calcineurin inhibitors. J. Alzheimers Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Hwang, J.Y.; Son, S.M.; Kim, Y.H.; Moon, M.; Inhee, M.J. FK506 reduces amyloid plaque burden and induces MMP-9 in AβPP/PS1 double transgenic mice. J. Alzheimers Dis. 2010, 22, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Nassal, D.; Gratz, D.; Hund, T.J. Challenges and opportunities for therapeutic targeting of calmodulin kinase in heart. Front. Pharmacol. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Khayatan, D.; Razavi, S.M.; Arab, Z.N.; Niknejad, A.H.; Nouri, K.; Momtaz, S.; Gumpricht, E.; Jamialahmadi, T.; Abdolghaffari, A.H.; Barreto, G.E.; et al. Protective effects of curcumin against traumatic brain injury. Biomed. Pharmacother. 2022, 154, 113621. [Google Scholar] [CrossRef] [PubMed]
- Fuloria, S.; Mehta, J.; Chandel, A.; Sekar, M.; Rani, N.N.I.M.; Begum, M.Y.; Subramaniyan, V.; Chidambaram, K.; Thangavelu, L.; Nordin, R.; et al. A Comprehensive Review on the Therapeutic Potential of Curcuma longa Linn. in Relation to its Major Active Constituent Curcumin. Front. Pharmacol. 2022, 13, 820806. [Google Scholar] [CrossRef]
- Voulgaropoulou, S.D.; van Amerlsvoort, T.A.M.; Prickaerts, J.; Vingerhoets, C. The effect of curcumin on cognition in Alzheimer’s disease and aging: A systematic review of pre-clinical and clinical studies. Brain Res. 2022, 1725, 14676–14690. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Day, D.H. Calcium and Non-Penetrating Traumatic Brain Injury: A Proposal for the Implementation of an Early Therapeutic Treatment for Initial Head Insults. Biomolecules 2024, 14, 853. https://doi.org/10.3390/biom14070853
O’Day DH. Calcium and Non-Penetrating Traumatic Brain Injury: A Proposal for the Implementation of an Early Therapeutic Treatment for Initial Head Insults. Biomolecules. 2024; 14(7):853. https://doi.org/10.3390/biom14070853
Chicago/Turabian StyleO’Day, Danton H. 2024. "Calcium and Non-Penetrating Traumatic Brain Injury: A Proposal for the Implementation of an Early Therapeutic Treatment for Initial Head Insults" Biomolecules 14, no. 7: 853. https://doi.org/10.3390/biom14070853
APA StyleO’Day, D. H. (2024). Calcium and Non-Penetrating Traumatic Brain Injury: A Proposal for the Implementation of an Early Therapeutic Treatment for Initial Head Insults. Biomolecules, 14(7), 853. https://doi.org/10.3390/biom14070853