
Towards a Treatment for Leukodystrophy Using Cell-Based Interception and Precision Medicine
 , , , , ,
, , , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cell-Based Interception and Precision Medicine Applied to Leukodystrophies
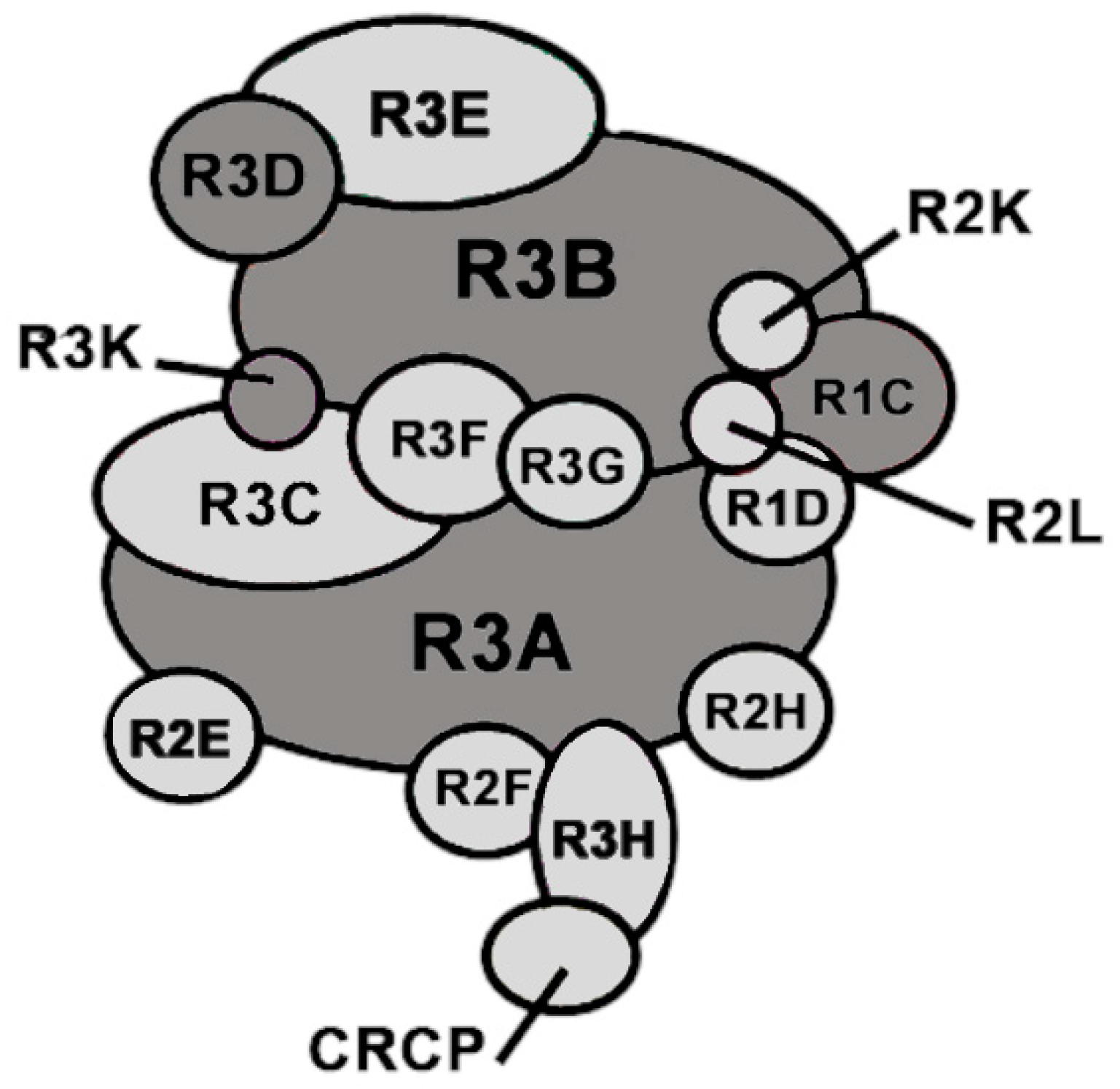
2.1. Detect
2.2. Understand
2.3. Therapy Development
2.4. Collaborate
3. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- LifeTime. Biomedical Research Initiative. Available online: https://lifetime-initiative.eu (accessed on 1 May 2024).
- Rajewsky, N.; Almouzni, G.; Gorski, S.A.; Aerts, S.; Amit, I.; Bertero, M.G.; Bock, C.; Bredenoord, A.L.; Cavalli, G.; Chiocca, S.; et al. LifeTime and improving European healthcare through cell-based interceptive medicine. Nature 2020, 587, 377–386. [Google Scholar] [CrossRef] [PubMed]
- 37TrilllionCells. Available online: https://37TrillionCells.com (accessed on 1 May 2024).
- Thiffault, I.; Wolf, N.I.; Forget, D.; Guerrero, K.; Tran, L.T.; Choquet, K.; Lavallee-Adam, M.; Poitras, C.; Brais, B.; Yoon, G.; et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun. 2015, 6, 7623. [Google Scholar] [CrossRef] [PubMed]
- Choquet, K.; Forget, D.; Meloche, E.; Dicaire, M.J.; Bernard, G.; Vanderver, A.; Schiffmann, R.; Fabian, M.R.; Teichmann, M.; Coulombe, B.; et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J. Biol. Chem. 2019, 294, 7445–7459. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.; Dastpeyman, S.; Poitras, C.; Bernard, G.; Gauthier, M.S.; Coulombe, B. Riluzole partially restores RNA polymerase III complex assembly in cells expressing the leukodystrophy-causative variant POLR3B R103H. Mol. Brain 2022, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.H.T.; Saksena, S.D.; Gifford, D.K. Generative modeling of single-cell time series with PRESCIENT enables prediction of cell trajectories with interventions. Nat. Commun. 2021, 12, 3222. [Google Scholar] [CrossRef]
- Vanderver, A.; Prust, M.; Tonduti, D.; Mochel, F.; Hussey, H.M.; Helman, G.; Garbern, J.; Eichler, F.; Labauge, P.; Aubourg, P.; et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 2015, 114, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; van der Knaap, M.S. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009, 72, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Steenweg, M.E.; Vanderver, A.; Blaser, S.; Bizzi, A.; de Koning, T.J.; Mancini, G.M.; van Wieringen, W.N.; Barkhof, F.; Wolf, N.I.; van der Knaap, M.S. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010, 133, 2971–2982. [Google Scholar] [CrossRef]
- Coulombe, B.; Derksen, A.; La Piana, R.; Brais, B.; Gauthier, M.S.; Bernard, G. POLR3-related leukodystrophy: How do mutations affecting RNA polymerase III subunits cause hypomyelination? Fac. Rev. 2021, 10, 12. [Google Scholar] [CrossRef]
- Adang, L.A.; Sherbini, O.; Ball, L.; Bloom, M.; Darbari, A.; Amartino, H.; DiVito, D.; Eichler, F.; Escolar, M.; Evans, S.H.; et al. Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol. Genet. Metab. 2017, 122, 18–32. [Google Scholar] [CrossRef]
- Keller, S.R.; Mallack, E.J.; Rubin, J.P.; Accardo, J.A.; Brault, J.A.; Corre, C.S.; Elizondo, C.; Garafola, J.; Jackson-Garcia, A.C.; Rhee, J.; et al. Practical Approaches and Knowledge Gaps in the Care for Children With Leukodystrophies. J. Child. Neurol. 2021, 36, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.; Chouery, E.; Putorti, M.L.; Tetreault, M.; Takanohashi, A.; Carosso, G.; Clement, I.; Boespflug-Tanguy, O.; Rodriguez, D.; Delague, V.; et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, M.; Choquet, K.; Orcesi, S.; Tonduti, D.; Balottin, U.; Teichmann, M.; Fribourg, S.; Schiffmann, R.; Brais, B.; Vanderver, A.; et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, J.; Perrier, S.; Pinard, M.; Tran, L.T.; Guerrero, K.; Prasad, C.; Prasad, A.N.; Pastinen, T.; Thiffault, I.; Coulombe, B.; et al. Biallelic pathogenic variants in POLR3D alter tRNA transcription and cause a hypomyelinating leukodystrophy: A case report. Front. Neurol. 2023, 14, 1254140. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.I.; Gutierrez Salazar, M.; Guerrero, K.; Thiffault, I.; Salomons, G.S.; Gauquelin, L.; Tran, L.T.; Forget, D.; Gauthier, M.S.; Waisfisz, Q.; et al. Bi-allelic Mutations in EPRS, Encoding the Glutamyl-Prolyl-Aminoacyl-tRNA Synthetase, Cause a Hypomyelinating Leukodystrophy. Am. J. Hum. Genet. 2018, 102, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Smith, D.E.; Issa, M.Y.; Stanley, V.; Wang, R.; Mendes, M.I.; Wright, M.S.; Wigby, K.; Hildreth, A.; Crawford, J.R.; et al. Biallelic mutations in valyl-tRNA synthetase gene VARS are associated with a progressive neurodevelopmental epileptic encephalopathy. Nat. Commun. 2019, 10, 707. [Google Scholar] [CrossRef] [PubMed]
- Lemire, G.; Ito, Y.A.; Marshall, A.E.; Chrestian, N.; Stanley, V.; Brady, L.; Tarnopolsky, M.; Curry, C.J.; Hartley, T.; Mears, W.; et al. ABHD16A deficiency causes a complicated form of hereditary spastic paraplegia associated with intellectual disability and cerebral anomalies. Am. J. Hum. Genet. 2021, 108, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Spahr, A.; Rosli, Z.; Legault, M.; Tran, L.T.; Fournier, S.; Toutounchi, H.; Darbelli, L.; Madjar, C.; Lucia, C.; St-Jean, M.L.; et al. The LORIS MyeliNeuroGene rare disease database for natural history studies and clinical trial readiness. Orphanet J. Rare Dis. 2021, 16, 328. [Google Scholar] [CrossRef] [PubMed]
- Soderholm, H.E.; Chapin, A.B.; Bayrak-Toydemir, P.; Bonkowsky, J.L. Elevated Leukodystrophy Incidence Predicted From Genomics Databases. Pediatr. Neurol. 2020, 111, 66–69. [Google Scholar] [CrossRef]
- Dorboz, I.; Dumay-Odelot, H.; Boussaid, K.; Bouyacoub, Y.; Barreau, P.; Samaan, S.; Jmel, H.; Eymard-Pierre, E.; Cances, C.; Bar, C.; et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol. Genet. 2018, 4, e289. [Google Scholar] [CrossRef]
- Wolf, N.I.; Vanderver, A.; van Spaendonk, R.M.; Schiffmann, R.; Brais, B.; Bugiani, M.; Sistermans, E.; Catsman-Berrevoets, C.; Kros, J.M.; Pinto, P.S.; et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 2014, 83, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Michell-Robinson, M.A.; Bernard, G. POLR3-Related Leukodystrophy: Exploring Potential Therapeutic Approaches. Front. Cell Neurosci. 2020, 14, 631802. [Google Scholar] [CrossRef] [PubMed]
- Choquet, K.; Pinard, M.; Yang, S.; Moir, R.D.; Poitras, C.; Dicaire, M.J.; Sgarioto, N.; Lariviere, R.; Kleinman, C.L.; Willis, I.M.; et al. The leukodystrophy mutation Polr3b R103H causes homozygote mouse embryonic lethality and impairs RNA polymerase III biogenesis. Mol. Brain 2019, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, E.P.; Abascal-Palacios, G.; Daiss, J.L.; King, H.; Gouge, J.; Pilsl, M.; Beuron, F.; Morris, E.; Gunkel, P.; Engel, C.; et al. Structure of human RNA polymerase III. Nat. Commun. 2020, 11, 6409. [Google Scholar] [CrossRef] [PubMed]
- Girbig, M.; Misiaszek, A.D.; Vorlander, M.K.; Lafita, A.; Grotsch, H.; Baudin, F.; Bateman, A.; Muller, C.W. Cryo-EM structures of human RNA polymerase III in its unbound and transcribing states. Nat. Struct. Mol. Biol. 2021, 28, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Vanderver, A.; Leventer, R.J.; Damiani, S.A.; Simons, C.; Grimmond, S.M.; Miller, D.; Schmidt, J.; Lockhart, P.J.; Pope, K.; et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 2013, 92, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Salomons, G.S.; Rodenburg, R.J.; Pouwels, P.J.; Schieving, J.H.; Derks, T.G.; Fock, J.M.; Rump, P.; van Beek, D.M.; van der Knaap, M.S.; et al. Mutations in RARS cause hypomyelination. Ann. Neurol. 2014, 76, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Lirussi, L.; Stromme, P.; Sumathipala, D.; Guerin, A.; Wolf, N.I.; Server, A.; Stensland, M.; Dalhus, B.; Tolun, A.; et al. A homozygous POLR1A variant causes leukodystrophy and affects protein homeostasis. Brain 2023, 146, 3513–3527. [Google Scholar] [CrossRef] [PubMed]
- Kara, B.; Koroglu, C.; Peltonen, K.; Steinberg, R.C.; Maras Genc, H.; Holtta-Vuori, M.; Guven, A.; Kanerva, K.; Kotil, T.; Solakoglu, S.; et al. Severe neurodegenerative disease in brothers with homozygous mutation in POLR1A. Eur. J. Hum. Genet. 2017, 25, 315–323. [Google Scholar] [CrossRef]
- Perrier, S.; Gauquelin, L.; Fallet-Bianco, C.; Dishop, M.K.; Michell-Robinson, M.A.; Tran, L.T.; Guerrero, K.; Darbelli, L.; Srour, M.; Petrecca, K.; et al. Expanding the phenotypic and molecular spectrum of RNA polymerase III-related leukodystrophy. Neurol. Genet. 2020, 6, e425. [Google Scholar] [CrossRef]
- Perrier, S.; Gauquelin, L.; Wambach, J.A.; Bernard, G. Distinguishing severe phenotypes associated with pathogenic variants in POLR3A. Am. J. Med. Genet. A 2022, 188, 708–712. [Google Scholar] [CrossRef] [PubMed]
- DeGasperis, S.M.; Bernard, G.; Wolf, N.I.; Miller, E.; Pohl, D. 4H leukodystrophy: Mild clinical phenotype and comorbidity with multiple sclerosis. Neurol. Genet. 2020, 6, e409. [Google Scholar] [CrossRef] [PubMed]
- Michell-Robinson, M.A.; Watt, K.E.N.; Grouza, V.; Macintosh, J.; Pinard, M.; Tuznik, M.; Chen, X.; Darbelli, L.; Wu, C.L.; Perrier, S.; et al. Hypomyelination, hypodontia and craniofacial abnormalities in a Polr3b mouse model of leukodystrophy. Brain 2023, 146, 5070–5085. [Google Scholar] [CrossRef] [PubMed]
- Merheb, E.; Cui, M.H.; DuBois, J.C.; Branch, C.A.; Gulinello, M.; Shafit-Zagardo, B.; Moir, R.D.; Willis, I.M. Defective myelination in an RNA polymerase III mutant leukodystrophic mouse. Proc. Natl. Acad. Sci. USA 2021, 118, e2024378118. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, J.; Michell-Robinson, M.; Chen, X.; Bernard, G. Decreased RNA polymerase III subunit expression leads to defects in oligodendrocyte development. Front. Neurosci. 2023, 17, 1167047. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef] [PubMed]
- Petelski, A.A.; Emmott, E.; Leduc, A.; Huffman, R.G.; Specht, H.; Perlman, D.H.; Slavov, N. Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 2021, 16, 5398–5425. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 50. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Huffman, R.G.; Chen, A.; Specht, H.; Slavov, N. DO-MS: Data-Driven Optimization of Mass Spectrometry Methods. J. Proteome Res. 2019, 18, 2493–2500. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Principal component analysis. Nat. Methods 2017, 14, 641–642. [Google Scholar] [CrossRef]
- Sangster, M.; Shahriar, S.; Niziolek, Z.; Carisi, M.C.; Lewandowski, M.; Budnik, B.; Grishchuk, Y. Brain cell type specific proteomics approach to discover pathological mechanisms in the childhood CNS disorder mucolipidosis type IV. Front. Mol. Neurosci. 2023, 16, 1215425. [Google Scholar] [CrossRef] [PubMed]
- Piscopo, V.E.C.; Chapleau, A.; Blaszczyk, G.J.; Sirois, J.; You, Z.; Soubannier, V.; Chen, C.X.; Bernard, G.; Antel, J.P.; Durcan, T.M. The use of a SOX10 reporter toward ameliorating oligodendrocyte lineage differentiation from human induced pluripotent stem cells. Glia 2024, 72, 1165–1182. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Fossati, V. Generation and isolation of oligodendrocyte progenitor cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 1143–1154. [Google Scholar] [CrossRef]
- Thomas, R.A.; Sirois, J.; Li, S.; Gestin, A.; Piscopo, V.E.; Lépine, P.; Mathur, M.; Chen, C.X.; Soubannier, V.; Goldsmith, T.M.; et al. Fon. CelltypeR: A flow cytometry pipeline to annotate, characterize and isolate single cells from brain organoids. BioRxiv 2023. [Google Scholar] [CrossRef]
- Chamling, X.; Kallman, A.; Fang, W.; Berlinicke, C.A.; Mertz, J.L.; Devkota, P.; Pantoja, I.E.M.; Smith, M.D.; Ji, Z.; Chang, C.; et al. Single-cell transcriptomic reveals molecular diversity and developmental heterogeneity of human stem cell-derived oligodendrocyte lineage cells. Nat. Commun. 2021, 12, 652. [Google Scholar] [CrossRef] [PubMed]
- Frazel, P.W.; Labib, D.; Fisher, T.; Brosh, R.; Pirjanian, N.; Marchildon, A.; Boeke, J.D.; Fossati, V.; Liddelow, S.A. Longitudinal scRNA-seq analysis in mouse and human informs optimization of rapid mouse astrocyte differentiation protocols. Nat. Neurosci. 2023, 26, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Tu, C.; Wang, J.; Yu, Y.; Guo, X.; Sun, J.; Sun, J.; Cai, W.; Yang, Q.; Sun, T. Deciphering Oligodendrocyte Lineages in the Human Fetal Central Nervous System Using Single-Cell RNA Sequencing. Mol. Neurobiol. 2023, 61, 1737–1752. [Google Scholar] [CrossRef]
- Dennis, D.J.; Wang, B.S.; Karamboulas, K.; Kaplan, D.R.; Miller, F.D. Single-cell approaches define two groups of mammalian oligodendrocyte precursor cells and their evolution over developmental time. Stem Cell Rep. 2024, 19, 654–672. [Google Scholar] [CrossRef] [PubMed]
- Jakel, S.; Agirre, E.; Mendanha Falcao, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcao, A.; Xiao, L.; Li, H.; Haring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Valihrach, L.; Matusova, Z.; Zucha, D.; Klassen, R.; Benesova, S.; Abaffy, P.; Kubista, M.; Anderova, M. Recent advances in deciphering oligodendrocyte heterogeneity with single-cell transcriptomics. Front. Cell Neurosci. 2022, 16, 1025012. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; van Velthoven, C.T.J.; Kunst, M.; Zhang, M.; McMillen, D.; Lee, C.; Jung, W.; Goldy, J.; Abdelhak, A.; Aitken, M.; et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 2023, 624, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Clair, G.C.; Williams, S.M.; Feng, S.; Tsai, C.F.; Moore, R.J.; Chrisler, W.B.; Smith, R.D.; Kelly, R.T.; Pasa-Tolic, L.; et al. Three-dimensional feature matching improves coverage for single-cell proteomics based on ion mobility filtering. Cell Syst. 2022, 13, 426–434 e424. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, F.A.; Thielert, M.; Strauss, M.T.; Schweizer, L.; Ammar, C.; Madler, S.C.; Metousis, A.; Skowronek, P.; Wahle, M.; Madden, K.; et al. Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome. Nat. Methods 2023, 20, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Straubhaar, J.; D’Souza, A.; Niziolek, Z.; Budnik, B. Single cell proteomics analysis of drug response shows its potential as a drug discovery platform. Mol. Omics 2024, 20, 6–18. [Google Scholar] [CrossRef]
- de la Fuente, A.G.; Queiroz, R.M.L.; Ghosh, T.; McMurran, C.E.; Cubillos, J.F.; Bergles, D.E.; Fitzgerald, D.C.; Jones, C.A.; Lilley, K.S.; Glover, C.P.; et al. Changes in the Oligodendrocyte Progenitor Cell Proteome with Ageing. Mol. Cell Proteomics 2020, 19, 1281–1302. [Google Scholar] [CrossRef]
- Gargareta, V.I.; Reuschenbach, J.; Siems, S.B.; Sun, T.; Piepkorn, L.; Mangana, C.; Spate, E.; Goebbels, S.; Huitinga, I.; Mobius, W.; et al. Conservation and divergence of myelin proteome and oligodendrocyte transcriptome profiles between humans and mice. Elife 2022, 11, e77019. [Google Scholar] [CrossRef]
- Feng, L.; Chao, J.; Zhang, M.; Pacquing, E.; Hu, W.; Shi, Y. Developing a human iPSC-derived three-dimensional myelin spheroid platform for modeling myelin diseases. iScience 2023, 26, 108037. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Calamini, B.; Morimoto, R.I. Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr. Top. Med. Chem. 2012, 12, 2623–2640. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coulombe, B.; Chapleau, A.; Macintosh, J.; Durcan, T.M.; Poitras, C.; Moursli, Y.A.; Faubert, D.; Pinard, M.; Bernard, G. Towards a Treatment for Leukodystrophy Using Cell-Based Interception and Precision Medicine. Biomolecules 2024, 14, 857. https://doi.org/10.3390/biom14070857
Coulombe B, Chapleau A, Macintosh J, Durcan TM, Poitras C, Moursli YA, Faubert D, Pinard M, Bernard G. Towards a Treatment for Leukodystrophy Using Cell-Based Interception and Precision Medicine. Biomolecules. 2024; 14(7):857. https://doi.org/10.3390/biom14070857
Chicago/Turabian StyleCoulombe, Benoit, Alexandra Chapleau, Julia Macintosh, Thomas M. Durcan, Christian Poitras, Yena A. Moursli, Denis Faubert, Maxime Pinard, and Geneviève Bernard. 2024. "Towards a Treatment for Leukodystrophy Using Cell-Based Interception and Precision Medicine" Biomolecules 14, no. 7: 857. https://doi.org/10.3390/biom14070857