
Molybdenum’s Role as an Essential Element in Enzymes Catabolizing Redox Reactions: A Review

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Selected Molybdenum-Containing Oxidating Enzymes
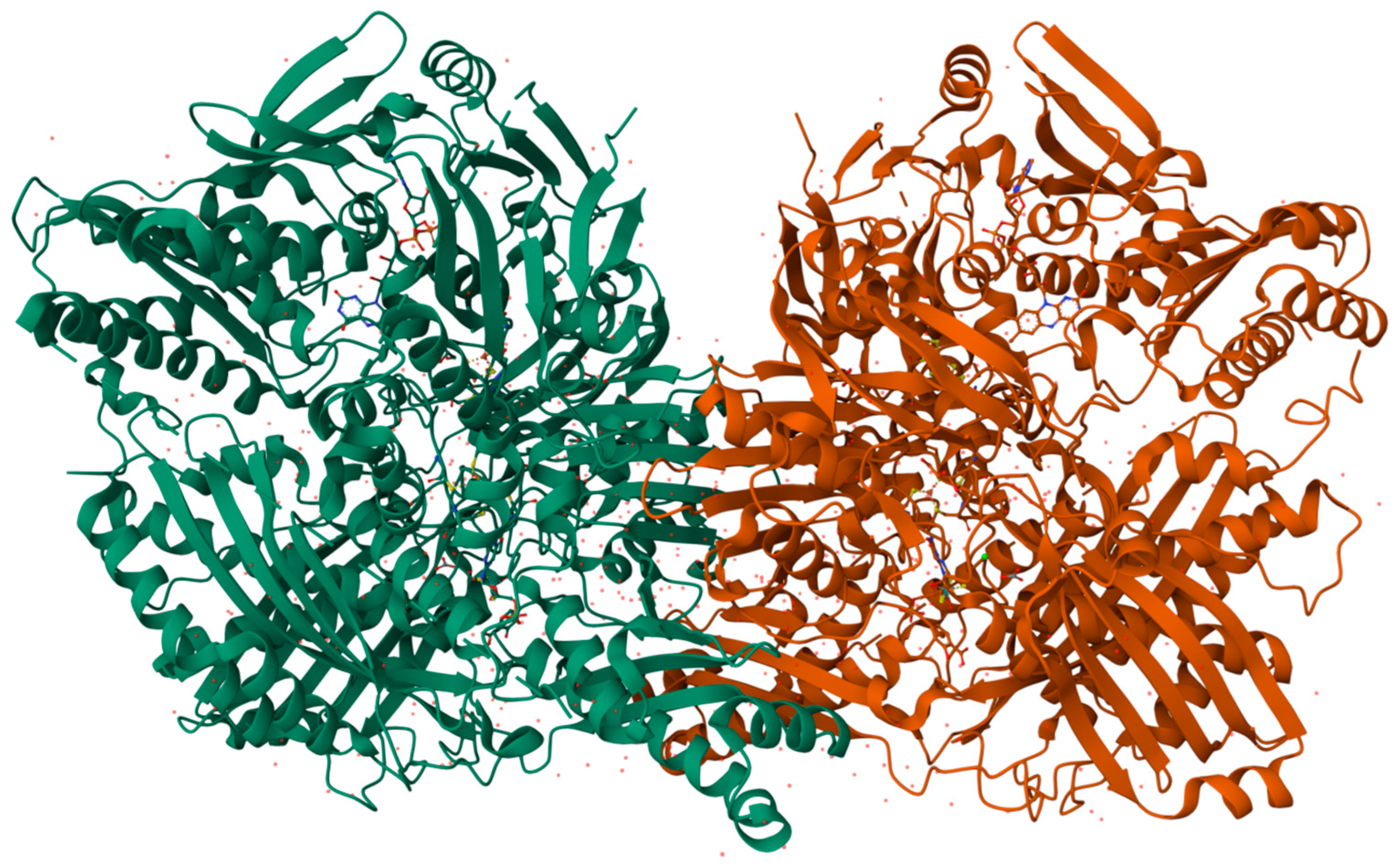
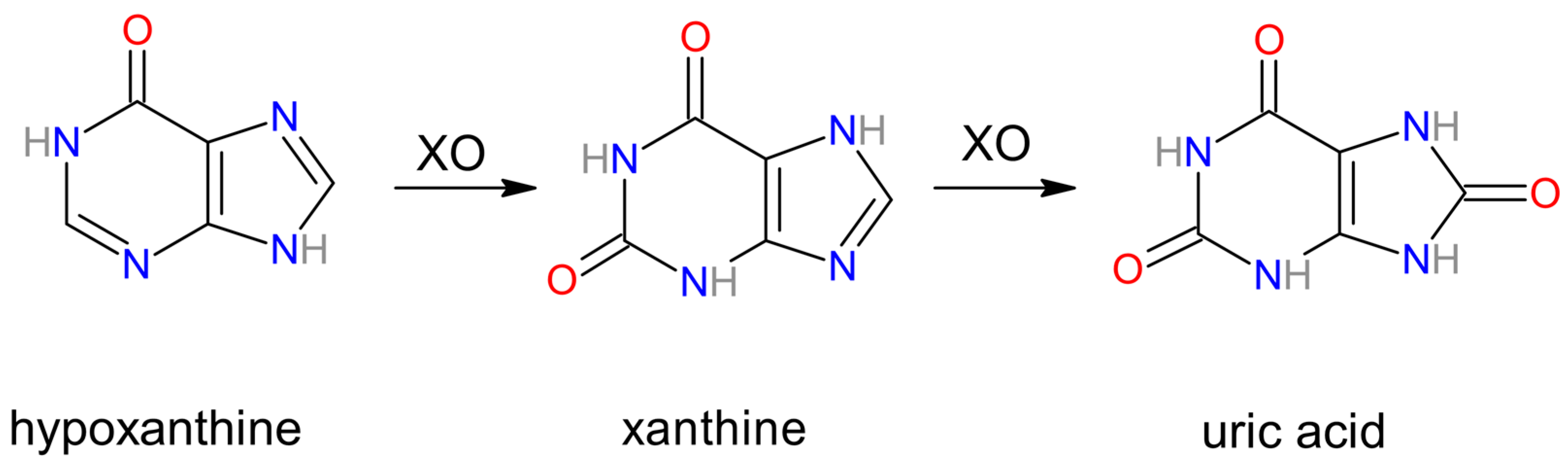
3.1. Xanthine Oxidase
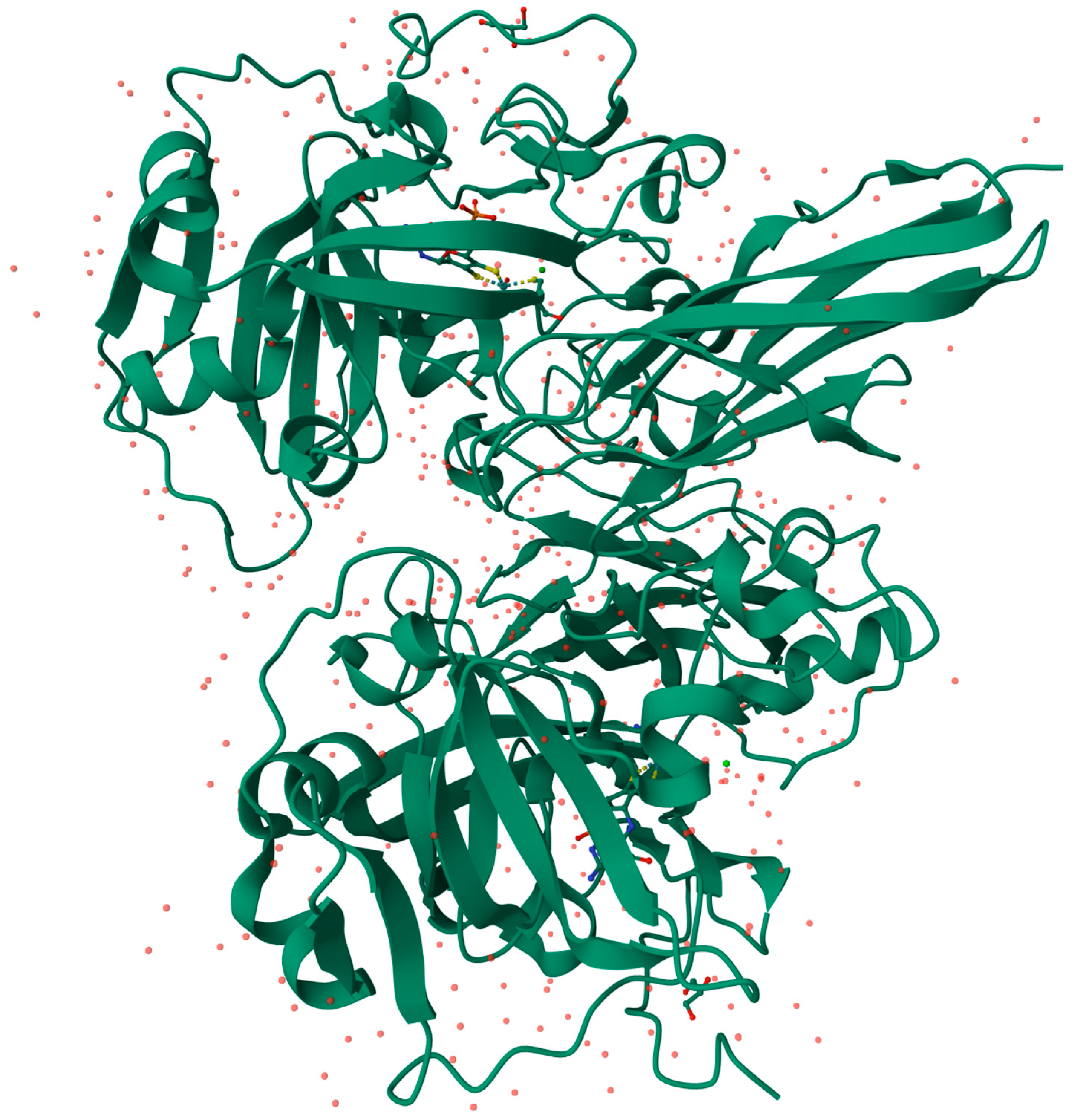
3.2. Aldehyde Oxidase
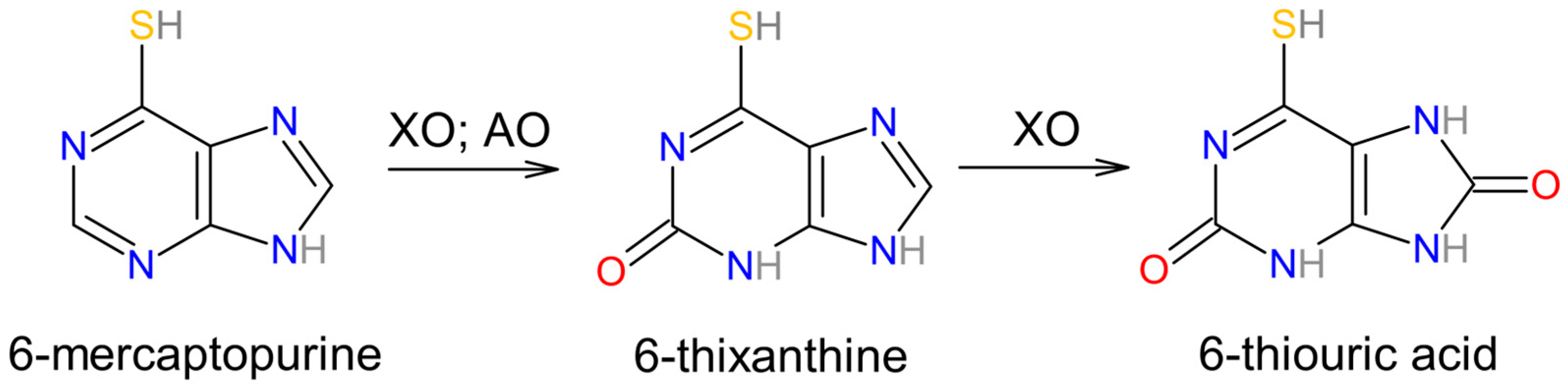
3.3. Involvement in Drug Metabolism of Aldehyde Oxidase and Xanthine Oxidase
3.4. Sulfite Oxidase
3.5. Sulfite Oxidase in Modern Clinical Use
3.6. Mitochondrial Amidoxime-Reducing Component
3.7. mARC as a Significant Component in Human Disease
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Novotny, J.A. Molybdenum Nutriture in Humans. J. Evid.-Based Complement. Altern. Med. 2011, 16, 164–168. [Google Scholar] [CrossRef]
- De Renzo, E.C.; Kaleita, E.; Heytler, P.G.; Oleson, J.J.; Hutchings, B.L.; Williams, J.H. Identification of the xanthine oxidase factor as molybdenum. Arch. Biochem. Biophys. 1953, 45, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Richert, D.A.; Westerfeld, W.W. Isolation and identification of the xanthine oxidase factor as molybdenum. J. Biol. Chem. 1953, 203, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Havemeyer, A.; Bittner, F.; Wollers, S.; Mendel, R.; Kunze, T.; Clement, B. Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J. Biol. Chem. 2006, 281, 34796–34802. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Struwe, M.A. The History of mARC. Molecules 2023, 28, 4713. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Alfaro, J.F.; Jones, J.P. Studies on the mechanism of aldehyde oxidase and xanthine oxidase. J. Org. Chem. 2008, 73, 9469–9472. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hille, R. Molybdenum-containing hydroxylases. Arch. Biochem. Biophys. 2005, 433, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Tollin, G.; Enemark, J.H. Sulfite oxidizing enzymes. Biochim. Biophys. Acta 2007, 1774, 527–539. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schroeder, H.A.; Balassa, J.J.; Tipton, I.H. Essential Trace Metals in Man: Molybdenum. J. Chronic Dis. 1970, 23, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Rosoff, B.; Spencer, H. The distribution and excretion of molybdenum-99 in mice. Health Phys. 1973, 25, 173–175. [Google Scholar] [PubMed]
- Tsongas, T.A.; Meglen, R.R.; Walravens, P.A.; Chappell, W.R. Molybdenum in the diet: An estimate of average daily intake in the United States. Am. J. Clin. Nutr. 1980, 33, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Novotny, J.A.; Turnlund, J.R. Molybdenum intake influences molybdenum kinetics in men. J. Nutr. 2007, 137, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Fu, C.Y.; Schwarz, G. Molybdenum Cofactor Deficiency in Humans. Molecules 2022, 27, 6896. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Johnson, J.L.; Duran, M. The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3163–3177. [Google Scholar]
- Adachi, T.; Fukushima, T.; Usami, Y.; Hirano, K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem. J. 1993, 289 Pt 2, 523–527. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jarasch, E.D.; Bruder, G.; Heid, H.W. Significance of xanthine oxidase in capillary endothelial cells. Acta Physiol. Scand. Suppl. 1986, 548, 39–46. [Google Scholar] [PubMed]
- Parks, D.A.; Williams, T.K.; Beckman, J.S. Conversion of xanthine dehydrogenase to oxidase in ischemic rat intestine: A reevaluation. Am. J. Physiol. 1988, 254 Pt 1, G768–G774. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Rutili, G.; McCord, J.M. Superoxide radicals in feline intestinal ischemia. Gastroenterology 1981, 81, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, E. Proposed Molecular Mechanism for the Action of Molybdenum in Enzymes: Coupled Proton and Electron Transfer. Proc. Natl. Acad. Sci. USA 1973, 70, 988–992. [Google Scholar] [CrossRef]
- Pauff, J.M.; Cao, H.; Hille, R. Substrate Orientation and Catalysis at the Molybdenum Site in Xanthine Oxidase. Crystal Structures in complex with Xanthne and Lumazine. J. Biol. Chem. 2009, 284, 8760–8776. [Google Scholar] [CrossRef]
- Mendel, R.R. The Molybdenum Cofactor. J. Biol. Chem. 2013, 288, 13165–13172. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kusano, T.; Nishino, T. Chemical Nature and Reaction Mechanisms of the Molybdenum Cofactor of Xanthine Oxidoreductase. Curr. Pharm. Des. 2013, 19, 2606–2614. [Google Scholar] [CrossRef] [PubMed]
- Hille, R. Xanthine Oxidase-A Personal History. Molecules 2023, 28, 1921. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ishikita, H.; Eger, B.T.; Okamoto, K.; Nishino, T.; Pai, E.F. Protein conformational gating of enzymatic activity in xanthine oxidoreductase. J. Am. Chem. Soc. 2012, 134, 999–1009, Protein Data Bank (PDB) ID: 3AX7. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harrison, R. Physiological roles of xanthine oxidoreductase. Drug Metab. Rev. 2004, 36, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, N.J.; Alhomida, A.S.; Dutta, G.P.; Pandey, V.C. Antagonist effect of chloroquine and tumor necrosis factor on hepatic oxidative stress and antioxidant defense in normal and Plasmodium yoelii nigeriensis-infected mice. In Vivo 2002, 16, 67–70. [Google Scholar] [PubMed]
- Sanni, L.A.; Fu, S.; Dean, R.T.; Bloomfield, G.; Stocker, R.; Chaudhri, G.; Dinauer, M.C.; Hunt, N.H. Are reactive oxygen species involved in the pathogenesis of murine cerebral malaria? J. Infect. Dis. 1999, 179, 217–222. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2022. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (accessed on 23 March 2024).
- Iwalokun, B.A.; Bamiro, S.B.; Ogunledun, A. Levels and interactions of plasma xanthine oxidase, catalase and liver function parameters in Nigerian children with Plasmodium falciparum infection. APMIS 2006, 114, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Musiani, S.; Valgimigli, M.; Gramantieri, L.; Tomassoni, F.; Bolondi, L.; Stirpe, F. Serum xanthine oxidase in human liver disease. Am. J. Gastroenterol. 2001, 96, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Rej, R. Aspartate aminotransferase activity and isoenzyme proportions in human liver tissues. Clin. Chem. 1978, 24, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Cholongitas, E.; Pavlopoulou, I.; Papatheodoridi, M.; Markakis, G.E.; Bouras, E.; Haidich, A.B.; Papatheodoridis, G. Epidemiology of nonalcoholic fatty liver disease in Europe: A systematic review and meta-analysis. Ann. Gastroenterol. 2021, 34, 404–414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.W.; Sung, K.C.; Shin, H.S.; Kim, B.J.; Kim, B.S.; Kang, J.H.; Lee, M.H.; Park, J.R.; Kim, H.; Rhee, E.J.; et al. Relationship between serum uric acid concentration and insulin resistance and metabolic syndrome. Circ. J. 2005, 69, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, C.; Wan, X.; Xu, L.; Weng, H.; Yan, M.; Miao, M.; Sun, Y.; Xu, G.; Dooley, S.; Li, Y.; et al. Xanthine oxidase in non-alcoholic fatty liver disease and hyperuricemia: One stone hits two birds. J. Hepatol. 2015, 62, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, R.; Wyndham, R.A.; Henry, N.P. On Liver Aldehydrase. Aust. J. Exp. Biol. Med. Sci. 1936, 14, 259–274. [Google Scholar] [CrossRef]
- Gordon, A.H.; Green, D.E.; Subrahmanyan, V. Liver aldehyde oxidase. Biochem. J. 1940, 34, 764–774. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Obach, R.S.; Huynh, P.; Allen, M.C.; Beedham, C. Human. liver aldehyde oxidase: Inhibition by 239 drugs. J. Clin. Pharmacol. 2004, 44, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Pubmed Search Results. Available online: https://pubmed.ncbi.nlm.nih.gov/?term=aldehyde+oxidase (accessed on 7 April 2024).
- Pryde, D.C.; Dalvie, D.; Hu, Q.; Jones, P.; Obach, R.S.; Tran, T.D. Aldehyde oxidase: An enzyme of emerging importance in drug discovery. J. Med. Chem. 2010, 53, 8441–8460. [Google Scholar] [CrossRef] [PubMed]
- Garattini, E.; Fratelli, M.; Terao, M. Mammalian aldehyde oxidases: Genetics, evolution and biochemistry. Cell Mol. Life Sci. 2008, 65, 1019–1048. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R. Cell biology of molybdenum. Biofactors 2009, 35, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Krenitsky, T.A.; Neil, S.M.; Elion, G.B.; Hitchings, G.H. A comparison of the specificities of xanthine oxidase and aldehyde oxidase. Arch. Biochem. Biophys. 1972, 150, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Sugihara, K.; Ohta, S. Drug-metabolizing ability of molybdenum hydroxylases. Drug Metab. Pharmacokinet. 2006, 21, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Beedham, C. Molybdenum Hydroxylases. In Enzyme Systems That Metabolise Drugs and Other Xenobiotics; Ioannides, C., Ed.; John Wiley and Sons: London, UK, 2001; pp. 147–187. [Google Scholar]
- Beedham, C. Molybdenum hydroxylases as drug-metabolizing enzymes. Drug Metab. Rev. 1985, 16, 119–156. [Google Scholar] [CrossRef] [PubMed]
- Coelho, C.; Foti, A.; Hartmann, T.; Santos-Silva, T.; Leimkühler, S.; Romão, M.J. Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat. Chem. Biol. 2015, 11, 779–783, Protein Data Bank (PDB) ID: 4UHW. [Google Scholar] [CrossRef] [PubMed]
- Beedham, C.; Bruce, S.E.; Critchley, D.J.; al-Tayib, Y.; Rance, D.J. Species variation in hepatic aldehyde oxidase activity. Eur. J. Drug Metab. Pharmacokinet. 1987, 12, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, Y.; Yamamoto, T.; Takahashi, S.; Tsutsumi, Z.; Hada, T. Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol. Histopathol. 2001, 16, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Garattini, E.; Terao, M. The role of aldehyde oxidase in drug metabolism. Expert. Opin. Drug Metab. Toxicol. 2012, 8, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Di, L.; Han, X.; Soderstrom, C.; Snyder, M.; Troutman, M.D.; Obach, S.R.; Zhang, H. Aldehyde Oxidase 1 (AOX1) in Human Liver Cytosols: Quantitative Characterization of AOX1 Expression Level and Activity Relationship. Drug Metab. Dispos. 2013, 41, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Choughule, K.V.; Barnaba, C.; Joswig-Jones, C.A.; Jones, J.P. In vitro oxidative metabolism of 6-mercaptopurine in human liver: Insights into the role of the molybdoflavoenzymes aldehyde oxidase, xanthine oxidase, and xanthine dehydrogenase. Drug Metab. Dispos. 2014, 42, 1334–1340. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burchenal, J.H.; Murphy, M.L.; Ellison, R.R.; Sykes, M.P.; Tan, T.C.; Leone, L.A.; Karnofsky, D.A.; Craver, L.F.; Dargeon, H.W.; Rhoads, C.P. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood 1953, 8, 965–999. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Pawa, S.; Naime, M.; Prasad, R.; Ahmad, T.; Farooqui, H.; Zafar, H. Role of mammalian cytosolic molybdenum Fe-S flavin hydroxylases in hepatic injury. Life Sci. 2008, 82, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Rajagopalan, K.V. Purification and properties of sulfite oxidase from human liver. J. Clin. Investig. 1976, 58, 543–550. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karakas, E.; Wilson, H.L.; Graf, T.N.; Xiang, S.; Jaramillo-Busquets, S.; Rajagopalan, K.V.; Kisker, C. Structural insights into sulfite oxidase deficiency. J. Biol. Chem. 2005, 280, 33506–33515, Protein Data Bank (PDB) ID: 2A99. [Google Scholar] [CrossRef] [PubMed]
- Kisker, C.; Schindelin, H.; Pacheco, A.; Wehbi, W.A.; Garrett, R.M.; Rajagopalan, K.V.; Enemark, J.H.; Rees, D.C. Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell 1997, 91, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Kappler, U.; Bailey, S. Molecular basis of intramolecular electron transfer in sulfite-oxidizing enzymes is revealed by high resolution structure of a heterodimeric complex of the catalytic molybdopterin subunit and a c-type cytochrome subunit. J. Biol. Chem. 2005, 280, 24999–25007. [Google Scholar] [CrossRef] [PubMed]
- Schrader, N.; Fischer, K.; Theis, K.; Mendel, R.R.; Schwarz, G.; Kisker, C. The crystal structure of plant sulfite oxidase provides insights into sulfite oxidation in plants and animals. Structure 2003, 11, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.Z.; Yu, W.L.; Dong, H.; Zhou, W.P.; Gu, Y.J.; Yu, H.; Yu, H.; Lu, X.Y.; Xian, Z.H.; Liu, Y.K.; et al. SUOX is a promising diagnostic and prognostic biomarker for hepatocellular carcinoma. J. Hepatol. 2013, 59, 510–517. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Projections of Mortality and Causes of Death, 2016 to 2060. Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 15 July 2024).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, E.; Colombo, M. Distinctive features of hepatocellular carcinoma in non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2016, 1, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Samant, H.; Amiri, H.S.; Zibari, G.B. Addressing the worldwide hepatocellular carcinoma: Epidemiology, prevention and management. J. Gastrointest. Oncol. 2021, 12 (Suppl. S2), S361–S373. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Di Tommaso, L.; Franchi, G.; Park, Y.N.; Fiamengo, B.; Destro, A.; Morenghi, E.; Montorsi, M.; Torzilli, G.; Tommasini, M.; Terracciano, L.; et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 2007, 45, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, L.; Destro, A.; Seok, J.Y.; Balladore, E.; Terracciano, L.; Sangiovanni, A.; Iavarone, M.; Colombo, M.; Jang, J.J.; Yu, E.; et al. The application of markers (HSP70 GPC3 and GS) in liver biopsies is useful for detection of hepatocellular carcinoma. J. Hepatol. 2009, 50, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Satow, R.; Shitashige, M.; Kanai, Y.; Takeshita, F.; Ojima, H.; Jigami, T.; Honda, K.; Kosuge, T.; Ochiya, T.; Hirohashi, S.; et al. Combined functional genome survey of therapeutic targets for hepatocellular carcinoma. Clin. Cancer Res. 2010, 16, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.Z.; Li, Y.; Cong, W.M.; Yu, H.; Dong, H.; Shu, H.; Liu, X.H.; Yan, G.Q.; Zhang, L.; Zhang, Y.; et al. iTRAQ-2DLC-ESI-MS/MS based identification of a new set of immunohistochemical biomarkers for classification of dysplastic nodules and small hepatocellular carcinoma. J. Proteome Res. 2011, 10, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, L.; Desmet, V.; Roskams, T. Preneoplastic lesions in human hepatocarcinogenesis. Liver Int. 2005, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.M.; Zondervan, P.E.; IJzermans, J.N.; Schalm, S.W.; de Man, R.A.; Krestin, G.P. Benign versus malignant hepatic nodules: MR imaging findings with pathologic correlation. Radiographics 2002, 22, 1023–1036; discussion 1037–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Vincent, A.S.; Halliwell, B.; Wong, K.P. A mechanism of sulfite neurotoxicity: Direct inhibition of glutamate dehydrogenase. J. Biol. Chem. 2004, 279, 43035–43045. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.H.; Eichler, F.S.; Hoda, S.; Lee, M.S.; Baris, H.; Hanley, C.A.; Grant, P.E.; Krishnamoorthy, K.S.; Shih, V.E. Isolated sulfite oxidase deficiency: A case report with a novel mutation and review of the literature. Pediatrics 2005, 116, 757–766, Erratum in Pediatrics 2005, 116, 1615. [Google Scholar] [CrossRef] [PubMed]
- Kocamaz, E.; Adiguzel, E.; Er, B.; Gundogdu, G.; Kucukatay, V. Sulfite leads to neuron loss in the hippocampus of both normal and SOX-deficient rats. Neurochem. Int. 2012, 61, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Christie, I.N.; Theparambil, S.M.; Braga, A.; Doronin, M.; Hosford, P.S.; Brazhe, A.; Mascarenhas, A.; Nizari, S.; Hadjihambi, A.; Wells, J.A.; et al. Astrocytes produce nitric oxide via nitrite reduction in mitochondria to regulate cerebral blood flow during brain hypoxia. Cell Rep. 2023, 42, 113514. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Plitzko, B.; Ott, G.; Reichmann, D.; Henderson, C.J.; Wolf, C.R.; Mendel, R.; Bittner, F.; Clement, B.; Havemeyer, A. The involvement of mitochondrial amidoxime reducing components 1 and 2 and mitochondrial cytochrome b5 in N-reductive metabolism in human cells. J. Biol. Chem. 2013, 288, 20228–20237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ott, G.; Havemeyer, A.; Clement, B. The mammalian molybdenum enzymes of mARC. J. Biol. Inorg. Chem. 2015, 20, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Wahl, B.; Reichmann, D.; Niks, D.; Krompholz, N.; Havemeyer, A.; Clement, B.; Messerschmidt, T.; Rothkegel, M.; Biester, H.; Hille, R.; et al. Biochemical and spectroscopic characterization of the human mitochondrial amidoxime reducing components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J. Biol. Chem. 2010, 285, 37847–37859. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gruenewald, S.; Wahl, B.; Bittner, F.; Hungeling, H.; Kanzow, S.; Kotthaus, J.; Schwering, U.; Mendel, R.R.; Clement, B. The fourth molybdenum containing enzyme mARC: Cloning and involvement in the activation of N-hydroxylated prodrugs. J. Med. Chem. 2008, 51, 8173–8177. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.W.T.; Ooi, J.S.G.; Joly, G.L.C.; Chan, K.R. Gene Updater: A web tool that autocorrects and updates for Excel misidentified gene names. Sci. Rep. 2022, 12, 12743. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kubitza, C.; Bittner, F.; Ginsel, C.; Havemeyer, A.; Clement, B.; Scheidig, A.J. Crystal structure of human mARC1 reveals its exceptional position among eukaryotic molybdenum enzymes. Proc. Natl. Acad. Sci. USA 2018, 115, 11958–11963, Protein Data Bank (PDB) ID: 6FW2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bauch, E.; Reichmann, D.; Mendel, R.R.; Bittner, F.; Manke, A.M.; Kurz, P.; Girreser, U.; Havemeyer, A.; Clement, B. Electrochemical and mARC-catalyzed enzymatic reduction of para-substituted benzamidoximes: Consequences for the prodrug concept “amidoximes instead of amidines”. ChemMedChem 2015, 10, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Havemeyer, A.; Grünewald, S.; Wahl, B.; Bittner, F.; Mendel, R.; Erdélyi, P.; Fischer, J.; Clement, B. Reduction of N-hydroxy-sulfonamides, including N-hydroxy-valdecoxib, by the molybdenum-containing enzyme mARC. Drug Metab. Dispos. 2010, 38, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Ginsel, C.; Plitzko, B.; Froriep, D.; Stolfa, D.A.; Jung, M.; Kubitza, C.; Scheidig, A.J.; Havemeyer, A.; Clement, B. The Involvement of the Mitochondrial Amidoxime Reducing Component (mARC) in the Reductive Metabolism of Hydroxamic Acids. Drug Metab. Dispos. 2018, 46, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Kotthaus, J.; Wahl, B.; Havemeyer, A.; Kotthaus, J.; Schade, D.; Garbe-Schönberg, D.; Mendel, R.; Bittner, F.; Clement, B. Reduction of N(ω)-hydroxy-L-arginine by the mitochondrial amidoxime reducing component (mARC). Biochem. J. 2011, 433, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Indorf, P.; Kubitza, C.; Scheidig, A.J.; Kunze, T.; Clement, B. Drug Metabolism by the Mitochondrial Amidoxime Reducing Component (mARC): Rapid Assay and Identification of New Substrates. J. Med. Chem. 2020, 63, 6538–6546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kozmin, S.G.; Leroy, P.; Pavlov, Y.I.; Schaaper, R.M. YcbX and yiiM, two novel determinants for resistance of Escherichia coli to N-hydroxylated base analogues. Mol. Microbiol. 2008, 68, 51–65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Plitzko, B.; Havemeyer, A.; Kunze, T.; Clement, B. The pivotal role of the mitochondrial amidoxime reducing component 2 in protecting human cells against apoptotic effects of the base analog N6-hydroxylaminopurine. J. Biol. Chem. 2015, 290, 10126–10135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krompholz, N.; Krischkowski, C.; Reichmann, D.; Garbe-Schönberg, D.; Mendel, R.R.; Bittner, F.; Clement, B.; Havemeyer, A. The mitochondrial Amidoxime Reducing Component (mARC) is involved in detoxification of N-hydroxylated base analogues. Chem. Res. Toxicol. 2012, 25, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, Y.; Yang, G.; Zhang, S.; Liu, Y.; Zhou, S.; Guo, H.; Liang, S.; Cui, Y.; Zhang, B.; et al. A novel mitochondrial amidoxime reducing component 2 is a favorable indicator of cancer and suppresses the progression of hepatocellular carcinoma by regulating the expression of p27. Oncogene 2020, 39, 6099–6112. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, D.; Liang, S.; Guo, H.; Zhang, S.; Yang, G.; Yuan, Y.; Liu, L. Downregulation of MARC2 Promotes Immune Escape and Is Associated with Immunosuppression of Hepatocellular Carcinoma. Front. Genet. 2022, 12, 790093. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Emdin, C.A.; Haas, M.E.; Khera, A.V.; Aragam, K.; Chaffin, M.; Klarin, D.; Hindy, G.; Jiang, L.; Wei, W.Q.; Feng, Q.; et al. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020, 16, e1008629, Erratum in PLoS Genet. 2021, 17, e1009503. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Innes, H.; Buch, S.; Hutchinson, S.; Guha, I.N.; Morling, J.R.; Barnes, E.; Irving, W.; Forrest, E.; Pedergnana, V.; Goldberg, D.; et al. Genome-Wide Association Study for Alcohol-Related Cirrhosis Identifies Risk Loci in MARC1 and HNRNPUL1. Gastroenterology 2020, 159, 1276–1289.e7. [Google Scholar] [CrossRef] [PubMed]
- Fairfield, C.J.; Drake, T.M.; Pius, R.; Bretherick, A.D.; Campbell, A.; Clark, D.W.; Fallowfield, J.A.; Hayward, C.; Henderson, N.C.; Joshi, P.K.; et al. Genome-Wide Association Study of NAFLD Using Electronic Health Records. Hepatol. Commun. 2022, 6, 297–308. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lewis, L.C.; Chen, L.; Hameed, L.S.; Kitchen, R.R.; Maroteau, C.; Nagarajan, S.R.; Norlin, J.; Daly, C.E.; Szczerbinska, I.; Hjuler, S.T.; et al. Hepatocyte mARC1 promotes fatty liver disease. JHEP Rep. 2023, 5, 100693. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamus, J.P.; Ruszczyńska, A.; Wyczałkowska-Tomasik, A. Molybdenum’s Role as an Essential Element in Enzymes Catabolizing Redox Reactions: A Review. Biomolecules 2024, 14, 869. https://doi.org/10.3390/biom14070869
Adamus JP, Ruszczyńska A, Wyczałkowska-Tomasik A. Molybdenum’s Role as an Essential Element in Enzymes Catabolizing Redox Reactions: A Review. Biomolecules. 2024; 14(7):869. https://doi.org/10.3390/biom14070869
Chicago/Turabian StyleAdamus, Jakub Piotr, Anna Ruszczyńska, and Aleksandra Wyczałkowska-Tomasik. 2024. "Molybdenum’s Role as an Essential Element in Enzymes Catabolizing Redox Reactions: A Review" Biomolecules 14, no. 7: 869. https://doi.org/10.3390/biom14070869