Abstract
When a genetic disease is characterized by the abnormal activation of normal molecular pathways and cellular events, it is illuminating to critically examine the places and times of these activities both in health and disease. Therefore, because heterotopic ossification (HO) in fibrodysplasia ossificans progressiva (FOP) is by far the disease’s most prominent symptom, attention is also directed toward the pathways and processes of bone formation during skeletal development. FOP is recognizable by effects of the causative mutation on skeletal development even before HO manifests, specifically in the malformation of the great toes. This signature skeletal phenotype is the most highly penetrant, but is only one among several skeletal abnormalities associated with FOP. Patients may present clinically with joint malformation and ankylosis, particularly in the cervical spine and costovertebral joints, as well as characteristic facial features and a litany of less common, non-skeletal symptoms, all stemming from missense mutations in the ACVR1 gene. In the same way that studying the genetic cause of HO advanced our understanding of HO initiation and progression, insight into the roles of ACVR1 signaling during tissue development, particularly in the musculoskeletal system, can be gained from examining altered skeletal development in individuals with FOP. This review will detail what is known about the molecular mechanisms of developmental phenotypes in FOP and the early role of ACVR1 in skeletal patterning and growth, as well as highlight how better understanding these processes may serve to advance patient care, assessments of patient outcomes, and the fields of bone and joint biology.
1. Introduction
Fibrodysplasia ossificans progressiva (FOP; MIM#135100) is a genetic disease characterized by the progressive immobilization of the body due to heterotopic ossification (HO) that restricts the function of joints [1,2]. Although HO in FOP is not detected prenatally, after birth, HO typically forms following the activation of the immune system through injury, vaccination, or illness, or spontaneously in an event of high, localized immune activity called a flare-up [3,4]. HO in FOP occurs through endochondral ossification (EO), similar to that of bone formation in most of the developing embryonic skeleton. It is distinct, however, in that EO during early development is tightly regulated and not associated with immune cell activity or inflammation, whereas HO grows irregularly and is commonly associated with both immune activity and inflammation [5,6,7]. Mature HO is distinct from normal bone in its aberrant morphology and mineral density, but histological examination reveals chondrocytes, osteoblasts, osteocytes, calcified tissue, and vasculogenesis—all hallmarks of healthy bone [5]. When expanding HO abuts primary skeletal structures, it may even form pseudo-joints, or systems in which dense tissue in the HO mass articulates with the skeleton and causes a mobile, painful, joint-like system. Together, these features support the assertion that the HO of FOP recapitulates many, but not all, of the developmental processes of EO in the skeleton, despite being distinct in its causation, timing, and location.
FOP has long been associated with characteristic skeletal malformations, particularly those of the great toe [8]. Therefore, we look to the endogenous skeleton for clues as to how the normal processes and pathways of bone formation could be co-opted elsewhere in the body by FOP. Skeletal maturation is dependent on bone morphogenetic proteins (BMPs), which were first identified and named for the ability to induce the formation of ectopic bone [9]. To regulate its potent morphogenetic ability, BMP pathway signaling is carefully coordinated among myriad extracellular and intracellular activators and inhibitors. Because of this bone-forming capacity, components of the BMP signaling pathway were among the first targets queried in molecular studies of the pathogenesis of FOP [10]. Among these is the receptor culpable for FOP, activin A receptor type I (ACVR1), also called activin receptor-like kinase-2 (ALK2) [11]. ACVR1 is a type I BMP receptor that, in unaffected individuals, helps regulate the initiation of an intracellular cascade to activate the genetic program of EO [12]. At sites of BMP pathway activity during embryonic skeletal formation, skeletal progenitor cells condense and differentiate into chondroblasts [13]. These chondroblasts mature, adjust their extracellular environment, and recruit additional cells to complete the metamorphosis of soft tissue into mineralized, vascularized bone. Therefore, the location and timing of BMP activity in the presence of cells capable of responding to it is of paramount importance. Otherwise, EO would proceed directionless and unrestricted, preventing the formation of the joints, tendons, and ligaments that allow the skeleton to move.
This review will detail the molecular, cellular, and tissue-level events of skeletal morphogenesis and how these are disrupted in the early development of individuals with FOP. It will subsequently discuss post-natal manifestations of enhanced BMP signaling through the mutant ACVR1 receptor. Finally, we will examine the impact of these phenotypes on patients and what that may mean for the discovery and assessment of current and future therapies for treating this devastating disease.
2. Molecular Basis of FOP
To understand how FOP-causing mutations disrupt signaling in various contexts, we will first review the BMP signaling pathway and its role in the endochondral ossification of the long bones of the limbs. Finer details of BMP pathways and the broader TGF-ß superfamily have been comprehensively reviewed elsewhere [14,15,16].
The initiation of canonical BMP pathway signaling requires the formation of a heterotetrameric complex composed of two type I and two type II transmembrane BMP receptors. These bind an extracellular dimer of two BMP ligands [17]. The formation of this complex promotes receptor conformational changes inside the cell to destabilize binding with the intracellular inhibitory protein FKBP12 and allow the serine/threonine kinase domain of the type II receptors to phosphorylate the glycine/serine-rich (GS) domain of the type I receptors [18]. This, in turn, phosphorylates the intracellular signal transducer complex, SMAD1/5/(8/9), which prompts the recruitment of the chaperone protein co-SMAD4 and the translocation of the active SMAD complex to the nucleus. This signal transduction via pSMAD1/5/(8/9) is called the canonical BMP pathway. Once it is in the nucleus, binding to BMP-responsive elements (BMEs) drives the transcription of target genes, including SOX9, a master regulator of chondrogenesis [19]. BMP signaling complexes may also phosphorylate TAK1 to activate the non-canonical p38 mitogen-activated protein kinase (MAPK) pathway and drive the transcription of RUNX2, DLX5, and SP7, leading to osteogenesis [15].
The assembly of various ligands and receptors into this membrane-bound complex and the subsequent selection of intracellular transducers—whether pSMAD1/5/(8/9) for the canonical BMP pathway, TAK1 for the non-canonical BMP pathway, or pSMAD2/3 for the TGFß pathway—is what permits the precise spatiotemporal modulation of BMP pathway activation [20,21,22]. Complexes containing overlapping constituent proteins may activate or inhibit the BMP pathway or activate the TGF-ß pathway [22]. Because these pathways are multimodal and present in a vast variety of developmental contexts, the absence or mutation of individual members may yield broad or specific consequences.
We consider these functions now in the context of the normal development of long bones, such as those of the limbs. In this process, canonical BMP pathway activation in condensations of mesenchymal cells causes endochondral ossification in embryonic tissues [13,23,24]. Between these condensations, BMP signaling is inhibited by Noggin, which permits the development of joint tissues [25]. These condensations undergo chondrogenesis in a stepwise fashion. First, BMP signaling and the subsequent expression of its direct transcriptional target SOX9 begin a transcriptional cascade, driving the differentiation of these cells into chondroblasts [19]. Chondroblasts proliferate and become polarized according to local directional molecular and physical cues. As these nascent chondroblasts stiffen the matrix around them with collagen, increasingly hypoxic conditions cue vasculogenesis through HIF1α activity and VEGF signaling. BMP signaling causes chondrocytes in the center of the cell mass to undergo hypertrophy and apoptosis or transdifferentiate to osteoblasts, which begin mineralizing the matrix around them. The newly stiffened and mineralized ECM plays a critical role in mesenchymal cell fate determination, with stiffer substrates promoting differentiation into osteoblasts and osteocytes [26].
As these bone anlagen progressively elongate, cells near the ends become organized in distinct layers. BMP is tightly controlled in a zonal fashion among these layers to allow other signals, including Wnt, PTHRP, and IHH, to maintain layers of articular cartilage at the epiphyses of the bone and layers of proliferating cells in the growth plates, which permit smoothly articulating joints and the longitudinal growth of bones, respectively [20,27]. The disruption of these balanced signals can diminish bone growth, lead to the premature loss and closure of growth plates, and prevent the formation and maintenance of articular cartilage [28,29]. Thus, healthy bone development is dependent on the regulation of BMP signaling, mechanotransduction, and hypoxia, all of which are implicated in models of FOP [30,31,32].
The processes and pathways described above are critically disrupted by FOP-causing mutations in the ACVR1 gene. The ACVR1 receptor is a type I BMP receptor typically found in canonical BMP-pathway-activating complexes [33,34]. It partners with other type I receptors, either BMPR1A/ALK3 or BMPR1B/ALK6 [35], and the type II receptors BMPR2 and ACVR2a [36,37]. It is the preferred target of BMP6 and BMP7 ligands and is also activated by BMP2 and BMP4 [17]. Most FOP patients share the same ACVR1 mutation: the replacement of arginine with histidine at position 206 (ACVR1-R206H) [11]. This and several other rarer pathogenic mutations cluster either within the GS domain, near the FKBP12-binding region, or within the protein kinase domain [38,39]. These non-R206H mutations are referred to as “variant” cases. Crystallography suggests R206 and Q207 stabilize FKBP12 binding to the GS domain [40] and that the R206H mutation reduces the stability of the FKBP12-ACVR1 complex compared to wild-type protein, which permits “leaky” signaling [41]. Experiments in mammalian cells show a reduced requirement of the BMP ligand for canonical pathway activation by ACVR1-R206H and the hyperactivation of the pathway in the presence of ligands [30]. The mutant receptor also shows reduced responsiveness to canonical inhibitors of signaling such as Noggin [42,43]. Experiments in zebrafish show a reduced requirement for partner type I receptors and the ligand binding of both classic and variant FOP receptors [44]. This reduced receptor requirement is accompanied by changes in GS domain activation requirements, permitting ACVR1-R206H to activate the SMAD complex more readily [44,45]. Together, these data show that ACVR1-R206H effectively raises the basal rate of BMP pathway activation and bypasses regulation by usual molecular mechanisms.
FOP-causing mutations also alter the ACVR1 receptor’s response to BMP/TGFß family ligands. BMP2, 4, 6, and 7 typically strongly activate the canonical BMP signaling pathway through ACVR1. In cells expressing either the recurrent R206H mutation or one of several variant FOP-causing mutations, BMP4 treatment yields a much higher phosphorylation of SMAD1/5/(8/9) [46]. Additional cell culture experiments revealed the enhanced responsiveness of ACVR1-R206H to BMP2, 4, 6, and 7 in transfected C2C12 cells [42], and to BMP6 and 7 in patient-derived induced pluripotent stem cells [37]. The mutations can also change context-dependent functions of certain ligands. Activin A binding to ACVR1-containing tetramers can act as a BMP pathway inhibitor by locking receptors into non-signaling complexes [47,48]. However, in the presence of the ACVR1-R206H mutation, Activin A strongly promotes BMP pathway activity, and blocking Activin antibodies in the presence of the FOP mutation decreases the incidence of trauma-induced HO [48,49,50,51,52]. The suppression of HO formation by palovarotene, an RARγ agonist, in individuals with FOP is accompanied by a reduced expression of Inhba, the gene encoding Activin A, further supporting a role for Activin A in the molecular pathogenesis of FOP [53].
The canonical transducer of BMP signaling, pSMAD1/5/(8/9), is shared with the RhoA pathway, which is a mechanotransduction pathway important for regulating cells’ movements and how they sense their environments [54,55]. By aberrantly phosphorylating SMAD, FOP mutations alter cells’ ability to sense and respond to physical forces in the microenvironment due to the crosstalk between these two pathways [31,56]. This increases the nuclear translocation of the mechanosensing pathway proteins YAP and TAZ, the expression of cartilage- and bone-associated genes, and the consequent differentiation of mesenchymal cells towards cartilage and bone fates [31,56].
In hypoxic conditions, BMP signaling complexes are retained in active states in endosomes. Cells with ACVR1-R206H mutant receptors retain these hypoxia-dependent endosomes longer than those with wild-type ACVR1, thereby extending the duration of enhanced BMP signaling [32]. Because bone-forming environments tend to be hypoxic and because the inhibition of HIF1α, a hypoxia-sensing protein, abrogates HO formation in models of FOP, HIF1α is a potent target for FOP therapies. Furthermore, the mechanistic target of rapamycin (mTOR) acts upstream of HIF1α. Targeting mTOR has been successful in human cell models and mouse models in reducing HO [57,58] and is an important signaling molecule in joint disease and maintenance, making it a promising candidate for tackling both HO and the non-HO symptoms of FOP. These two factors have recently been reviewed elsewhere [59].
3. The Great Toe of FOP
One of the most curious features of FOP is also one of the most important for providing insight into how the disease affects skeletal biology and the role of ACVR1 in early development: the bilateral malformation of the great toes [8]. At birth, most individuals with FOP are noted to have unusual, laterally deviated great toes. This characteristic FOP malformation is typically referred to as hallux valgus with absent or malformed joints. All affected individuals have malformation of the distal metatarsal, while half of cases show a loss of one phalanx, and approximately one-third show a longitudinal epiphyseal bracket (a condition almost never observed in the hallux except in FOP), in which the growth plate extends longitudinally along the phalanx [60]. FOP variant mutations may lead to much more severe digit phenotypes, including syndactyly and severe reduction in multiple digits in both the hands and feet [38]. These together suggest a disruption in the proximal–distal pre-patterning of the phalanges and begin to paint a picture of the molecular pathogenesis of the development of the normotopic skeleton of FOP.
In normal vertebrate development, digits of the hands and feet are patterned from proximal to distal according to a pattern of BMP pathway activity in which high activity yields a phalanx, metacarpal, or metatarsal, and low activity denotes a joint interzone (Figure 1 [21,61]. In humans, mice, and most related mammals, this pattern yields three phalanges in each of digits 2–5, and two phalanges in digit 1, which corresponds to both the thumb and the great toe. As with other long bones of the limbs, phalanges begin as primary ossification centers that grow circumferentially while lengthening along the proximo-distal axis, aided by growth plates containing rapidly dividing chondroprogenitor cells [62]. Physical or molecular disruptions may alter the pattern along this axis, thereby changing the number of joints or skeletal elements, or alter the direction of growth, leading to bunions or other altered morphology.
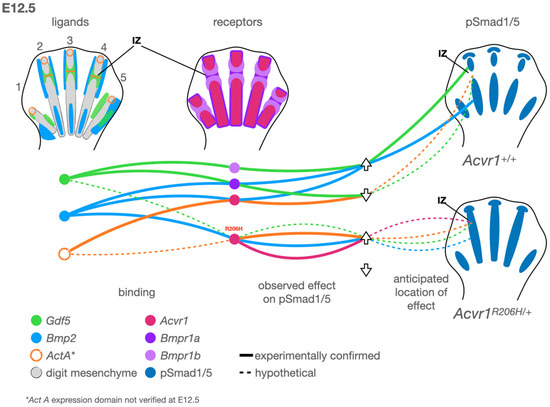
Figure 1.
A model for altered regulation of BMP pathway activity during digit development by Acvr1 R206H. Left: Observed expression patterns of the ligands Gdf5 (green), Bmp2 (light blue), and ActA (orange, based on data from chickens). Middle: Type 1 receptors Acvr1 (fuchsia), Bmpr1a (purple), and Bmpr1b (light purple). Right: Observed pSmad1/5 domains (dark blue) in control (top right) and Acvr1R206H/+ (bottom right) mouse limbs. At this early stage, digits 1 and 5 have not yet acquired the transverse stripes of Gdf5 expression associated with joint interzones (IZ). Arrows indicate whether the ligand and receptor connected by the associated line up- or down-regulates canonical BMP pathway activity via Smad1/5 phosphorylation. Solid lines indicate binding and activity observed experimentally. Dotted lines indicate hypotheses supported by altered Acvr1 activity in the presence of FOP-causing mutations. Gdf5 attenuates BMP-pSmad1/5 activity through Bmpr1a binding in a context-dependent manner, which is hypothesized (dotted line) to restrict BMP pathway activity at the interzone (IZ). In vitro, Bmp2-Acvr1 binding activates pSmad1/5, but ActA-Acvr1 binding reduces or nullifies pSmad1/5. Acvr1-R206H binds Bmp2 as a potent activator and cells expressing Acvr1-R206H strongly activate pSmad1/5 when treated with ActA. Acvr1-R206H can also signal ligand-independently. Based on expression patterns, receptor–ligand binding capabilities, and signaling through these complexes, we hypothesize the following: ligand-independent Acvr1-R206H activity contributes to multiple aspects of the observed phenotype; ActA aberrantly promotes IZ pSmad1/5 activity in a digit-independent manner; Gdf5-Acvr1 signaling promotes digit-specific IZ pSmad1/5 activity; and Bmp2-Acvr1-R206H likely contributes to persistent chondrogenic activity in the developing digit skeleton.
Great toe malformation is represented in genetic mouse models of FOP [63,64], providing a unique opportunity to study the etiology of this phenomenon. However, Acvr1 is necessary to complete gastrulation in mice, and the global expression of the R206H mutation in mice leads to perinatal lethality [64,65,66], necessitating that experimental systems restrict knock-out and knock-in alleles to specific cell lineages. Acvr1 is expressed at low levels during early murine skeletogenesis and is rapidly restricted to expression in the periosteum, the cell layer surrounding the hardened cortical bone in long bones like those of the limbs [12,67]. Mice lacking Acvr1 expression in Col2a1+ cells show a global delay in chondrogenesis and a delay in osteogenesis in the digits [12]. Mice lacking Acvr1 expression in Prrx1+ osteochondroprogenitor cells show significant disruption of the first digit, with soft tissue connecting two proximal elements to an unidentified distal element [68]. Mice expressing the R206H mutation only in Prrx1+ cells have severe limb deformities [63,64] but show a particular reduction in skeletal elements, dysregulated growth plate polarity, and the loss of joint structures in the first digit of the hindlimb [67], closely mirroring the human FOP toe phenotype. Importantly, these mice also show malformations of the fifth digit, which is seen frequently in the hands of humans with FOP [8,69], but not the feet [60,69], suggesting a more general mechanism of action in the limb than a first-digit-specific effect. In mice surviving to four weeks of age, articular cartilage of the knees is significantly reduced. Though mice globally expressing the R206H mutation survive only a few hours after birth, they show a reduction in all limb skeletal elements, with the strongest effects on preaxial elements, i.e., the radius, fibula, and great toe [67]. These phenotypes were not present in mice expressing the mutation in only Scx+ (tenocytes) or Mx+ (bone-marrow-derived endochondral progenitors) cells [70]. Mice expressing the engineered, constitutively active mutant receptor Acvr1-Q207D typically do not survive gestation, though mice expressing that receptor only in Nfatc1+ cells showed HO in the wrists, ankles, and phalanges in association with joint tissues, as opposed to originating from within the skeletal muscle tissue [71]. The infection of chicken embryos with retrovirus containing the R206H, Q207D, or Q207E mutant receptors leads to fused joints in the limbs and digits [40], suggesting an evolutionarily conserved function for Acvr1 in joint and digit development. Together, these studies show that Acvr1 acts during skeletal patterning to regulate chondrogenesis, as well as the site-specific contributions of cells to joint structures.
Acvr1 clearly plays important roles in early skeletal patterning, particularly in the limbs, but the exact molecular mechanism or mechanisms of this contribution and how it achieves such remarkable specificity are unknown. In mammals, the first digit is transcriptionally distinct from the other four in that it is considered SHH-independent and only expresses one of the 5′ HOXD cluster indispensable for digit morphogenesis [72]. However, digit malformations in both humans and mice with FOP-causing mutations are not restricted to digit one. While this does not preclude a role for the first digit’s unique identity in the etiology of the FOP great toe phenotype, it suggests a more broadly acting mechanism. For instance, one role of BMP in the developing limb is to ultimately interrupt cell proliferation and drive differentiation; therefore, BMP activity is at first inhibited in the most distal cells to permit continued outgrowth. The disruption of that inhibition leads to premature chondrification and reduced digit length [73]. Mice expressing Acvr1-R206H show significantly reduced digit length and possible altered SMAD1/5/(8/9) in the distal digit tips, though the latter observation has not been quantified [67]. Another possible explanation lies in the molecular mechanism of determining the number, position, and size of skeletal elements in the hands and feet. These features are thought to be partially determined by Türing reaction–diffusion, in which a long-range initiator of a signal and a short-range inhibitor of that signal interact with local geometry over time to determine stable active and inactive zones [61]. BMP pathway activity and Sox9-driven chondrogenesis are thought to be positive/active outputs of this pathway, with regions of high BMP pathway activity driving the differentiation of the cartilage template for the skeleton, but the specific components and the results of small perturbations are not fully understood. Therefore, it may be that the altered responsiveness of the Acvr1-R206H receptor to stimuli and leaky signaling disrupt the normal generation and maintenance of this reaction–diffusion mechanism in such a way that only specific digits are affected. Variations in either BMP-regulated limb bud outgrowth or a BMP component of Türing reaction–diffusion should therefore produce a gradient of severities. Though a clean gradient of phenotypes has not been described, variant FOP-causing mutations can lead to a variety of distal limb phenotypes, including syndactyly, the loss of digits, the loss of all medial phalanges, and the loss of nail beds (i.e., disruption to the distal tips of digits) [38].
4. Altered Skeletal and Joint Development in FOP
The developmental skeletal phenotype of FOP appears to be primarily due to disrupted joint formation. Understanding the specifics of toe malformation can help to understand both its etiology and how it may be connected to other, less common developmental features of FOP (Figure 2). These include hip dysplasia [74,75], hand malformations [69,74,76], fusions of the cervical spine [76,77], distinct craniofacial features [78,79,80], fusions of the temporomandibular joint [79], metaphyseal osteochondromas [81], and exostosis-like mineralized tendon insertion sites [82]. Skeletal malformations in FOP must be distinguished between primary malformations caused by altered development and secondary deformities resulting from the presence of heterotopic bone or altered posture and gait [74,83]. In particular, the tibio-fibular joints, femoral necks, and cervical spine show fusion and dysmorphia in early life, which, in the absence of HO near the affected areas, suggests those symptoms have developmental origins [84]. Large-cohort natural history studies support that osteochondromas, osteophytes, fusions of spinal elements, joint degeneration, hip dysplasia, and intra-articular ankyloses of costovertebral joints may also occur in the absence of focal HO, suggesting that the FOP mutation drives degenerative symptoms separate from HO well after primary skeletal development has completed [74,84,85]. Pertinent to laboratory studies, genetic knock-in mouse models are true to these features of FOP: the global or mosaic knock-in of Acvr1-R206H can drive all of the aforementioned skeletal phenotypes [63]; Prrx1-specific expression can drive all limb phenotypes [64]; and the Sox10-specific expression of Acvr1-R206H or the P0-specific expression of Acvr1-Q207D can drive craniofacial phenotypes, the latter due to altered cranial neural crest cell migration [86,87]. Notably, the appearance of osteochondromas, intra-articular ankyloses, the exostosis-like mineralization of tendons, and osteophytes are all disorders involving the aberrant growth of bony tissue not necessarily in the context of injury. These suggest a more general ability of the FOP mutation to drive osteogenesis in multiple clinically relevant contexts, rather than only during flare-ups. While the precise impact of each of these phenotypes is uncertain, the non-HO characteristics of FOP contribute to a gradual loss of mobility, and those affecting the costovertebral joints are implicated in thoracic insufficiency syndrome, which is responsible for a high percentage of morbidity in FOP [88].
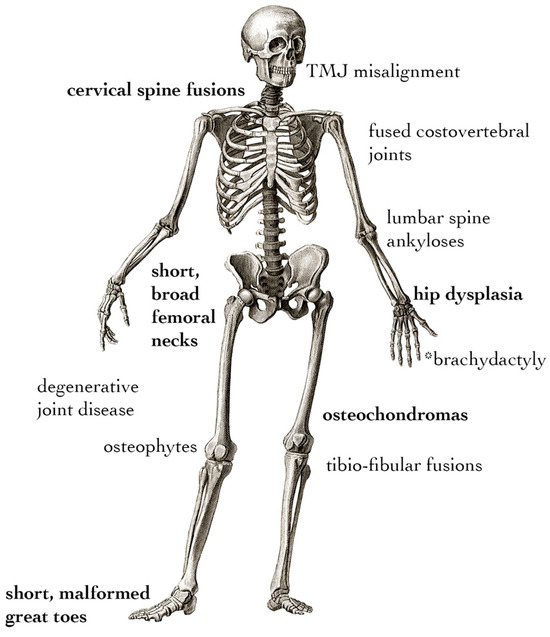
Figure 2.
Non-HO skeletal phenotypes of FOP. Symptoms in bold are present in the majority of individuals with FOP, whereas the remainder arise with variable penetration among patients. * Brachydactyly is only associated with non-R206H variant cases.
We will first consider the events of embryonic joint development before applying these concepts to that of altered digit and joint patterning in FOP (Figure 1). On a cellular level, presumptive skeletal joints are identified by low canonical BMP pathway activity, the expression of Gdf5 (a BMP pathway ligand), and the gradual migration of Gdf5+ cells into the joint interzone, the space directly between two adjacent bones (Figure 1) [89,90,91]. These cells are specified based on the timing of this migration, with early-migrating cells differentiating into tissues, including articular cartilage, and late-migrating cells contributing to tendons, ligaments, and synovium [92]. The disruption of Gdf5+ cell migration can lead to site-specific defects ranging from a failure of joint cavitation and interzone formation to a delayed or aberrant development of tendons, ligaments, and articular cartilage. The mutation of GDF5 is associated with multiple brachydactylies (OMIM #615072, #112600, and #113100) and appendicular dysplasias (OMIM #200700 and #228900). BMP must be inhibited at sites of joint formation to prevent the differentiation of cells to cartilage and to allow the joint to cavitate [20,21]. The Wnt pathway signaling, which often acts in opposition to the BMP pathway, must be active in the interzone [93]. The genetic deletion of BMP receptors [94] or members of the Wnt pathway [93] can lead to a failure of joint progenitors to migrate and differentiate in a site-specific manner. Activins, TGF-ß, and their associated receptors are present in the developing limb, and TGF-ß pathway signaling activity is required for skeletal development. However, the precise roles of these factors at the level of digit joint formation have not yet been untangled (reviewed in depth elsewhere [95]). The loss of Noggin, a primary BMP ligand antagonist, leads to a total loss of synovial joint formation in mice [25] and a spectrum of digit phenotypes in humans, including brachydactyly type B2 (OMIM #611377) and proximal symphalangism 1A (OMIM #185800). Thus, while the differential contributions of receptors, ligands, and resultant complexes may have joint-specific roles, BMP inhibition is absolutely required for proper joint development.
In situ hybridization experiments in Acvr1R206H/+; Prrx1-Cre mouse limbs suggest that Gdf5+ cells do not properly localize to the presumptive joint interzone in a digit-specific manner [67]. This altered localization is concurrent with aberrant pSMAD1/5 activity in the presumptive digit joint interzones and directly precedes defective skeletal and joint patterning in the digit. However, other joints develop mostly normally, matching observations in the FOP human patient population, suggesting the presentation is more complex than a generic loss of BMP pathway inhibition in all joints, as in Noggin knock-out models. The FOP mouse phenotype is more similar to Gdf5 homozygous knock-out [96], and an FOP-like great toe malformation has been reported in one patient deficient for BMPR1B [97] and another with a potentially causative point mutation in BMPR1B [98]. BMPR1B is a type I receptor that preferentially binds GDF5 ligands to drive joint formation, the loss of which is associated with human brachydactylies (OMIM #616849 and 112600) [99], suggesting that the FOP joint phenotype may be primarily, though not entirely, due to a disruption in GDF5 function. An intriguing possible explanation for this is that even though GDF5 can act as a context-specific chondrogenic factor by activating the canonical BMP pathway, it may instead act as a high-binding, low-activity sink for receptors in the joint interzone [100], an idea supported by GDF5’s typically weak activation across different receptors [22]; therefore, the constitutive activation of ACVR1 by the R206H mutation may circumvent such a function, leading to joint fusion in a pattern remarkably similar to GDF5 loss of function. In the brachydactylies referenced above, axial joints are typically unaffected, while appendicular joints are highly affected. Thus, the development of joints other than those in the digits may progress without defects, or with less severe defects that then become more apparent when challenged (reviewed elsewhere [101,102]), possibly explaining the early degenerative joint disease observed in FOP patients.
Changes in signaling in adult joint tissues that disrupt tissue maintenance and homeostasis can also presage degenerative osteoarthritis (OA), the appearance of osteochondromas, and the mineralization of cartilaginous connective tissues [103], all of which are observed at high rates and early ages in FOP compared to the general population [84]. The balance of BMP and TGF-ß signaling is critical in articular cartilage maintenance. One primary factor in OA is chondrocyte hypertrophy, a process normally driven by BMP pathway signaling during embryonic development. A mouse model of osteoarthritis shows the upregulation of the TGF-ß receptor Cripto, which participates in a BMP-pathway-activating complex [104]. The inhibition of Acvr1-BMP signaling has successfully reduced osteoarthritis progression in mice [105]. ACVR1 was also up-regulated in hypertrophic chondrocytes taken from the articular cartilage of osteoarthritis patients [106]. Together, these implicate enhanced basal BMP signaling as a candidate for the joint degeneration seen in people with FOP.
The localization of joint pathology in FOP suggests several factors acting individually or in concert at different sites in the body. One is altered interactions with locally expressed genes such as GDF5 during embryonic development and early childhood, thus mimicking the joint diseases usually associated with these genes. A second factor is minor alterations in embryonic and childhood joint development that are not initially observable but become apparent as the individual ages. A third is altered BMP pathway signaling within the mature joint that leads to premature joint degeneration. Finally, there are HO-caused changes in posture and gait, which can damage articular cartilage and other joint structures over time. Considering the variability in disease progression among individuals, it is likely that these combine uniquely depending on natural history.
5. Non-Skeletal Symptoms of FOP
Though FOP is primarily a disease of HO formation and altered skeletal development, other symptoms also arise with variable penetrance, especially in rare, variant cases in which ACVR1 mutations other than R206H are observed. These include alopecia, the loss of fingernails and toenails, and severe conductive hearing loss [38], as well as a suite of cardiopulmonary and neurologic phenotypes reviewed by Khan et al. [107]. The involvement of multiple organs apart from the skeletal system is in some ways to be expected because the BMP pathway is evolutionarily ancient and plays critical roles in morphogenesis throughout the body. Though specific molecular developmental mechanisms for the below listed phenotypes have not been thoroughly investigated, we may speculate on how ACVR1-R206H is able to manifest them.
- Alopecia: FOP patients with either classic or variant mutations may present with thinning or lost hair. The molecular development and maintenance of hair follicles relies on gene networks including BMP and Wnt signaling, with BMP promoting a quiescent state and Wnt promoting an active one [108]. The loss of hair-follicle-specific BMP signaling results in dysfunctional follicular morphogenesis [109]. The overexpression of Acvr1 in the hair follicle alters follicle morphology and localization, as well as wound healing [110]. Thus, hair thinning and hair loss in FOP are likely a primary effect of the hyperactivation of BMP pathway activity by ACVR1-R206H, though the effects of this specific mutation in this context have not been rigorously investigated.
- Loss of fingernails and toenails: Variant FOP mutations may also be associated with the loss of some or all fingernails and/or toenails. The nail bed is a densely cellular tissue with an active stem cell niche maintained by Wnt pathway signaling, whereas BMP is implicated in terminal nail cell differentiation [111]. The aberrant activation of BMP pathway signaling in the nail bed may deplete this stem cell population or prevent it from forming during early development. Further, nailbeds are features of the distal tip of the digits, and if digit patterning is so severely altered by pathogenic ACVR1 signaling as to stunt digit outgrowth, a niche for the nailbed might never be established.
- Hearing loss: The pathogenesis of hearing loss in FOP is symptomatically mostly conductive due to developmental fusion of the ossicles of the middle ear. However, some individuals with FOP have neural hearing impairment. The cochlea develops primarily under the control of Wnts, Fgfs, and Shh, but also requires a gradient of BMP pathway activity to refine sensory structures and aide in the development of hair cells [112]. While the expression of ACVR1 in sensory organs has not been detailed, the disruption of this gradient may lead to impaired neural hearing in some individuals with FOP.
- Cardiac phenotypes: Individuals with FOP have long been observed to have subclinical cardiac anomalies observed by electrocardiogram [113,114]. ACVR1 function is required for the development of multiple components of the heart such as the endocardial cushion [115,116]. Because of the known roles of ACVR1 in heart development, it is possible cardiac anomalies are primary symptoms of altered BMP pathway signaling; however, FOP also frequently restricts the chest wall, which may lead to changes in cardiopulmonary function as well.
- Neurologic dysfunctions: FOP is associated with a range of neurological symptoms [117]. Neuropathic pain, focal demyelination, and central nervous system patterning in general have all been linked to BMP pathway signaling. Mice expressing Acvr1-R206H have significant focal demyelination, and a clinical report showed multiple demyelination lesions in four FOP patients, though the demyelination could not be directly linked to neuropathy [118]. While the specific molecular mechanism has not been investigated, there are clear avenues for ACVR1 mutations to lead to multiple neurological defects.
6. FOP Treatments and Skeletal Development
Recently, several therapeutic approaches for treating FOP have shown promise in the prevention and abrogation of flare-ups and subsequent HO. While HO is the primary symptom contributing to reduced quality of life, other manifestations of the disease discussed here prompt a consideration of how treatments for FOP may impact the progression of phenotypes of the normotopic skeleton and joints. Clinical trials of FOP provide several unique challenges which have been thoroughly outlined [119]. Though HO is preferentially confirmed by computed tomography (CT), acquiring these data requires patients to assume positions that may be painful, dangerous, or impossible due to immobilizing HO. Such limitation also impacts the collection of data on joint and normotopic skeletal health. Therefore, patient- and clinician-reported outcomes must often be relied upon. In designing and assessing future clinical trials and the long-term outcomes of potential therapeutic agents, it will therefore be important to assess joint health and the potential retardation of developmental arthropathy as a viable clinical outcome independently of HO. Until then, investigators and clinicians, including the majority of trials referenced below, make use of the cumulative analogue joint involvement score (CAJIS). CAJIS was developed to be a snapshot of mobility burden in patients with FOP and assesses the impact of HO on joint function, but also provides inference into possible joint deterioration in the absence of reported or observed flare-ups and HO [120].
The following therapies are in recently completed, ongoing, or upcoming clinical trials and are detailed on the International FOP Association website (https://www.ifopa.org/ongoing_clinical_trials_in_fop; accessed on 24 June 2024) as of the publication of this article. While a thorough examination is beyond the scope of this review, we provide a brief consideration of how each relates to non-HO symptoms of FOP.
Palovarotene is a highly specific retinoic acid receptor gamma (RARγ) agonist that successfully abrogates cardiotoxin injury-induced HO in mouse models of FOP and appears to improve growth plate health, which is compromised by the Acvr1 R206H mutation [64,121]. However, concerns were raised early on about the skeletotoxicity of palovarotene in mouse models, which caused synovial joint hypertrophy followed by the premature closure of growth plates, leading to reduced growth and development [122]. This may be due to the off-target effects of the anti-chondrogenic properties of RAR agonists, which may deplete the stem cell pools in growth plates needed to continue longitudinal bone growth, and may implicate a delicate balance among Acvr1, RARγ, and other signaling pathways in growing bone. In clinical trials, the drug reduced both flare-up incidence and the progression of HO in patients but had side-effects including the premature closure of growth plates [123]. An upcoming long-term palovarotene study (NCT06089616) will include skeletal age, physeal closure, and height velocity as secondary outcomes, which may provide some insight into non-HO symptoms, though they may be conflated by HO lesions impinging on growth and joint function.
Rapamycin is a well-known inhibitor of mTOR kinase with immunosuppressant functions currently in clinical trials to treat FOP (UMIN000028429). As mentioned previously, mTOR and its regulation of HIF1α are potent targets for preventing traumatic and genetic HO [59], as well as their involvement in osteoarthritis and other degenerative joint diseases [124]. It will therefore be especially interesting to note whether rapamycin has any effect on non-HO-related changes to CAJIS.
Garetosmab, an activin A-blocking antibody, entered clinical trials [125] following successful reduction in HO in mouse models [49]. This treatment was not reported to have deleterious developmental defects; however, the mouse studies did not investigate the effects on growth plate closure or synovial joints. The current trial (NCT05394116) includes an assessment of patient joint function that may allude to degenerative joint disease but, like other trials, does not include specific indicators of joint disease as study parameters.
Recent investigations in the clinic and in FOP mouse models revealed that even the partial inhibition of matrix metalloproteinase-9 (MMP-9) activity by genetic, pharmacologic, or biologic means potently inhibits HO. Thus, it appears that MMP-9 is a vital molecular link between inflammation and HO in FOP, unveiling a novel treatment strategy for FOP [126,127].
A primary difficulty with pharmaceutical options for FOP has been the close sequence similarity between ACVR1 and other type I receptors, which creates high risk for side effects when ACVR1 is directly targeted. Modern small-molecule inhibitors either designed to or able to minimize off-target effects are now in various stages of clinical trials as well, including zilurgisertib (INCB000928), fidrisertib (IPN60130), and saracatinib (AZD0530). Due to their high specificity, these therapies may show promise for mitigating both HO and non-HO symptoms.
Adeno-associated virus (AAV)-based therapies have soared in use and pursuit in the past several years. Recently, Yang and colleagues published a study using AAV to suppress transcripts encoding ACVR1-R206H and produce healthy ACVR1 transcripts [128,129]. Encouragingly, this method showed decreased HO load in mice, as well as reductions in degenerative joint disease symptoms, vertebral fusions, and osteochondromas [128]. Though AAV therapies for FOP have not yet reached clinical trials, these preliminary investigations show exciting promise.
While HO correctly remains the primary target of potential FOP therapies, the growth plate health and joint tissue phenotypes that worsen with age represent a knowledge gap that should be considered during the assessment of clinical trial outcomes as new therapies are investigated. The nature of the disease precludes or makes onerous certain definitive measures of joint and skeletal health, however, so there is understandably a practical limit to the acquisition of such data.
7. Conclusions
“To measure the great toe of the foot is to measure the giant.”Victor Hugo
FOP is a complex disease despite its deceptively simple genetic origin: a single, recurrent base-pair substitution leading to a devastating outcome. While the burden of HO is certainly the most critical component of the disease to be addressed, our understanding of the molecular etiology of other symptoms associated with FOP has greatly improved in the past several years. Understanding the cellular and molecular signaling in a disease and looking deeply into seemingly minor phenotypes can yield a vastly improved comprehension of both fundamental biological processes and those of rare diseases, with significant improvements in quality of life. FOP is a disease not just of HO formation, but also of joint, skeletal, cardiac, and neurological development and maintenance. As exciting new therapies are developed and tested, it is important to understand the many different temporal and biological niches of BMP signaling to give patients the best possible information and outcomes.
Author Contributions
Writing and figures—O.W.T. Critique, revisions, and editing—E.M.S. and F.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
I would like to thank the FOP community for their inspirational interest in biomedical research that makes the search for cures possible.
Conflicts of Interest
F.S.K. is a founding member and past president of the International Clinical Council on FOP; a member of the Medical Advisory Board of the IFOPA Global Registry; and a global PI on the Regeneron Pharmaceuticals LUMINA-1 and the Clementia/Ipsen MOVE trials.
References
- Kaplan, F.S.; Smith, R.M. Fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 1997, 12, 855. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Shen, Q.; Lounev, V.; Seemann, P.; Groppe, J.; Katagiri, T.; Pignolo, R.J.; Shore, E.M. Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Metab. 2008, 26, 521–530. [Google Scholar] [CrossRef]
- Gannon, F.H.; Glaser, D.; Caron, R.; Thompson, L.D.; Shore, E.M.; Kaplan, F.S. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum. Pathol. 2001, 32, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Pignolo, R.J.; Shore, E.M. Granting immunity to FOP and catching heterotopic ossification in the Act. Semin. Cell Dev. Biol. 2016, 49, 30–36. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Tabas, J.A.; Gannon, F.H.; Finkel, G.; Hahn, G.V.; Zasloff, M.A. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J. Bone Jt. Surg. Am. 1993, 75, 220–230. [Google Scholar] [CrossRef]
- Convente, M.R.; Wang, H.; Pignolo, R.J.; Kaplan, F.S.; Shore, E.M. The immunological contribution to heterotopic ossification disorders. Curr. Osteoporos. Rep. 2015, 13, 116–124. [Google Scholar] [CrossRef]
- Convente, M.R.; Chakkalakal, S.A.; Yang, E.; Caron, R.J.; Zhang, D.; Kambayashi, T.; Kaplan, F.S.; Shore, E.M. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1(R206H) Mouse Model of Fibrodysplasia Ossificans Progressiva. J. Bone Miner. Res. 2018, 33, 269–282. [Google Scholar] [CrossRef]
- Connor, J.M.; Evans, D.A. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J. Bone Jt. Surg. Br. 1982, 64, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Virdi, A.S.; Shore, E.M.; Oreffo, R.O.; Li, M.; Connor, J.M.; Smith, R.; Kaplan, F.S.; Triffitt, J.T. Phenotypic and molecular heterogeneity in fibrodysplasia ossificans progressiva. Calcif. Tissue Int. 1999, 65, 250–255. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Rigueur, D.; Brugger, S.; Anbarchian, T.; Kim, J.K.; Lee, Y.; Lyons, K.M. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 2015, 30, 733–741. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Tsuji, K.; Cox, K.; Harfe, B.D.; Rosen, V.; Tabin, C.J. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006, 2, e216. [Google Scholar] [CrossRef] [PubMed]
- Zinski, J.; Tajer, B.; Mullins, M.C. TGF-beta Family Signaling in Early Vertebrate Development. Cold Spring Harb. Perspect. Biol. 2018, 10, a033274. [Google Scholar] [CrossRef]
- Koosha, E.; Eames, B.F. Two Modulators of Skeletal Development: BMPs and Proteoglycans. J. Dev. Biol. 2022, 10, 15. [Google Scholar] [CrossRef]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-beta signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.; Mueller, T.D. Specification of BMP Signaling. Cells 2019, 8, 1579. [Google Scholar] [CrossRef]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Behringer, R.R.; de Crombrugghe, B. L-Sox5, Sox6 and Sox9 control essential steps of the chondrocyte differentiation pathway. Osteoarthr. Cartil. 2001, 9 (Suppl. A), S69–S75. [Google Scholar] [CrossRef]
- Ray, A.; Singh, P.N.; Sohaskey, M.L.; Harland, R.M.; Bandyopadhyay, A. Precise spatial restriction of BMP signaling is essential for articular cartilage differentiation. Development 2015, 142, 1169–1179. [Google Scholar] [CrossRef]
- Singh, P.N.P.; Shea, C.A.; Sonker, S.K.; Rolfe, R.A.; Ray, A.; Kumar, S.; Gupta, P.; Murphy, P.; Bandyopadhyay, A. Precise spatial restriction of BMP signaling in developing joints is perturbed upon loss of embryo movement. Development 2018, 145, dev153460. [Google Scholar] [CrossRef] [PubMed]
- Klumpe, H.E.; Langley, M.A.; Linton, J.M.; Su, C.J.; Antebi, Y.E.; Elowitz, M.B. The context-dependent, combinatorial logic of BMP signaling. Cell Syst. 2022, 13, 388–407.e310. [Google Scholar] [CrossRef] [PubMed]
- Fell, H.B.; Robison, R. The growth, development and phosphatase activity of embryonic avian femora and limb-buds cultivated in vitro. Biochem. J. 1929, 23, 767–784.5. [Google Scholar] [CrossRef]
- Akiyama, H.; Lefebvre, V. Unraveling the transcriptional regulatory machinery in chondrogenesis. J. Bone Miner. Metab. 2011, 29, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Brunet, L.J.; McMahon, J.A.; McMahon, A.P.; Harland, R.M. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science 1998, 280, 1455–1457. [Google Scholar] [CrossRef]
- Tee, S.Y.; Fu, J.; Chen, C.S.; Janmey, P.A. Cell shape and substrate rigidity both regulate cell stiffness. Biophys. J. 2011, 100, L25–L27. [Google Scholar] [CrossRef]
- Amano, K.; Densmore, M.J.; Lanske, B. Conditional Deletion of Indian Hedgehog in Limb Mesenchyme Results in Complete Loss of Growth Plate Formation but Allows Mature Osteoblast Differentiation. J. Bone Miner. Res. 2015, 30, 2262–2272. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef]
- Chau, M.; Forcinito, P.; Andrade, A.C.; Hegde, A.; Ahn, S.; Lui, J.C.; Baron, J.; Nilsson, O. Organization of the Indian hedgehog--parathyroid hormone-related protein system in the postnatal growth plate. J. Mol. Endocrinol. 2011, 47, 99–107. [Google Scholar] [CrossRef]
- Culbert, A.L.; Chakkalakal, S.A.; Theosmy, E.G.; Brennan, T.A.; Kaplan, F.S.; Shore, E.M. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem. Cells 2014, 32, 1289–1300. [Google Scholar] [CrossRef]
- Stanley, A.; Heo, S.J.; Mauck, R.L.; Mourkioti, F.; Shore, E.M. Elevated BMP and Mechanical Signaling through YAP1/RhoA Poises FOP Mesenchymal Progenitors for Osteogenesis. J. Bone Miner. Res. 2019, 34, 1894–1909. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lindborg, C.; Lounev, V.; Kim, J.H.; McCarrick-Walmsley, R.; Xu, M.; Mangiavini, L.; Groppe, J.C.; Shore, E.M.; Schipani, E.; et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J. Bone Miner. Res. 2016, 31, 1652–1665. [Google Scholar] [CrossRef]
- Macias-Silva, M.; Hoodless, P.A.; Tang, S.J.; Buchwald, M.; Wrana, J.L. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 1998, 273, 25628–25636. [Google Scholar] [CrossRef]
- Zhang, D.; Schwarz, E.M.; Rosier, R.N.; Zuscik, M.J.; Puzas, J.E.; O’Keefe, R.J. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 2003, 18, 1593–1604. [Google Scholar] [CrossRef]
- Little, S.C.; Mullins, M.C. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis. Nat. Cell Biol. 2009, 11, 637–643. [Google Scholar] [CrossRef]
- Bagarova, J.; Vonner, A.J.; Armstrong, K.A.; Borgermann, J.; Lai, C.S.; Deng, D.Y.; Beppu, H.; Alfano, I.; Filippakopoulos, P.; Morrell, N.W.; et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol. Cell Biol. 2013, 33, 2413–2424. [Google Scholar] [CrossRef]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Jain, P.; Resnick, A.C. Shared ACVR1 mutations in FOP and DIPG: Opportunities and challenges in extending biological and clinical implications across rare diseases. Bone 2018, 109, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Deichsel, A.; Stange, K.; Ast, C.; Bocciardi, R.; Ravazzolo, R.; Di Rocco, M.; Ferrari, P.; Landi, A.; Kaplan, F.S.; et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum. Mol. Genet. 2014, 23, 5364–5377. [Google Scholar] [CrossRef]
- van Dinther, M.; Visser, N.; de Gorter, D.J.; Doorn, J.; Goumans, M.J.; de Boer, J.; ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Jones, W.D.; Hale, M.; Warder, B.N.; Shore, E.M.; Mullins, M.C. Reduced GS Domain Serine/Threonine Requirements of Fibrodysplasia Ossificans Progressiva Mutant Type I BMP Receptor ACVR1 in the Zebrafish. J. Bone Miner. Res. 2023, 38, 1364–1385. [Google Scholar] [CrossRef]
- Allen, R.S.; Tajer, B.; Shore, E.M.; Mullins, M.C. Fibrodysplasia ossificans progressiva mutant ACVR1 signals by multiple modalities in the developing zebrafish. Elife 2020, 9, 53761. [Google Scholar] [CrossRef]
- Haupt, J.; Xu, M.; Shore, E.M. Variable signaling activity by FOP ACVR1 mutations. Bone 2018, 109, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9, 54582. [Google Scholar] [CrossRef]
- Hildebrand, L.; Stange, K.; Deichsel, A.; Gossen, M.; Seemann, P. The Fibrodysplasia Ossificans Progressiva (FOP) mutation p.R206H in ACVR1 confers an altered ligand response. Cell Signal 2017, 29, 23–30. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Upadhyay, J.; Xie, L.; Huang, L.; Das, N.; Stewart, R.C.; Lyon, M.C.; Palmer, K.; Rajamani, S.; Graul, C.; Lobo, M.; et al. The Expansion of Heterotopic Bone in Fibrodysplasia Ossificans Progressiva Is Activin A-Dependent. J. Bone Miner. Res. 2017, 32, 2489–2499. [Google Scholar] [CrossRef]
- Wang, H.; Shore, E.M.; Pignolo, R.J.; Kaplan, F.S. Activin A amplifies dysregulated BMP signaling and induces chondro-osseous differentiation of primary connective tissue progenitor cells in patients with fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Mundy, C.; Yao, L.; Sinha, S.; Chung, J.; Rux, D.; Catheline, S.E.; Koyama, E.; Qin, L.; Pacifici, M. Activin A promotes the development of acquired heterotopic ossification and is an effective target for disease attenuation in mice. Sci. Signal 2021, 14, e10821. [Google Scholar] [CrossRef]
- Mundy, C.; Yao, L.; Shaughnessy, K.A.; Saunders, C.; Shore, E.M.; Koyama, E.; Pacifici, M. Palovarotene Action Against Heterotopic Ossification Includes a Reduction of Local Participating Activin A-Expressing Cell Populations. JBMR Plus 2023, 7, e10821. [Google Scholar] [CrossRef]
- Worthylake, R.A.; Burridge, K. RhoA and ROCK promote migration by limiting membrane protrusions. J. Biol. Chem. 2003, 278, 13578–13584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Y.; Gunst, S.J. The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J. Biol. Chem. 2012, 287, 33996–34008. [Google Scholar] [CrossRef]
- Haupt, J.; Stanley, A.; McLeod, C.M.; Cosgrove, B.D.; Culbert, A.L.; Wang, L.; Mourkioti, F.; Mauck, R.L.; Shore, E.M. ACVR1(R206H) FOP mutation alters mechanosensing and tissue stiffness during heterotopic ossification. Mol. Biol. Cell 2019, 30, 17–29. [Google Scholar] [CrossRef]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A.; et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef]
- Wang, H.; Kaplan, F.S.; Pignolo, R.J. The HIF-1alpha and mTOR Pathways Amplify Heterotopic Ossification. Biomolecules 2024, 14, 147. [Google Scholar] [CrossRef]
- Towler, O.W.; Kaplan, F.S.; Shore, E.M. The Developmental Phenotype of the Great Toe in Fibrodysplasia Ossificans Progressiva. Front. Cell Dev. Biol. 2020, 8, 612853. [Google Scholar] [CrossRef] [PubMed]
- Raspopovic, J.; Marcon, L.; Russo, L.; Sharpe, J. Modeling digits. Digit patterning is controlled by a Bmp-Sox9-Wnt Turing network modulated by morphogen gradients. Science 2014, 345, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Kelikian, A.S.; Sarrafian, S.K. Sarrafian’s Anatomy of the Foot and Ankle: Descriptive, Topographic, Functional; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Chakkalakal, S.A.; Zhang, D.; Culbert, A.L.; Convente, M.R.; Caron, R.J.; Wright, A.C.; Maidment, A.D.; Kaplan, F.S.; Shore, E.M. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012, 27, 1746–1756. [Google Scholar] [CrossRef]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef]
- Gu, Z.; Reynolds, E.M.; Song, J.; Lei, H.; Feijen, A.; Yu, L.; He, W.; MacLaughlin, D.T.; van den Eijnden-van Raaij, J.; Donahoe, P.K.; et al. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development 1999, 126, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Scott, G.; Nagy, A.; Kaartinen, V.; Mishina, Y. BMP type I receptor ALK2 is essential for proper patterning at late gastrulation during mouse embryogenesis. Dev. Dyn. 2007, 236, 512–517. [Google Scholar] [CrossRef]
- Towler, O.W.; Peck, S.H.; Kaplan, F.S.; Shore, E.M. Dysregulated BMP signaling through ACVR1 impairs digit joint development in fibrodysplasia ossificans progressiva (FOP). Dev. Biol. 2021, 470, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, L.; Schmidt-von Kegler, M.; Walther, M.; Seemann, P.; Stange, K. Limb specific Acvr1-knockout during embryogenesis in mice exhibits great toe malformation as seen in Fibrodysplasia Ossificans Progressiva (FOP). Dev. Dyn. 2019, 248, 396–403. [Google Scholar] [CrossRef]
- Schroeder, H.W., Jr.; Zasloff, M. The hand and foot malformations in fibrodysplasia ossificans progressiva. Johns Hopkins Med. J. 1980, 147, 73–78. [Google Scholar] [PubMed]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- Agarwal, S.; Loder, S.J.; Brownley, C.; Eboda, O.; Peterson, J.R.; Hayano, S.; Wu, B.; Zhao, B.; Kaartinen, V.; Wong, V.C.; et al. BMP signaling mediated by constitutively active Activin type 1 receptor (ACVR1) results in ectopic bone formation localized to distal extremity joints. Dev. Biol. 2015, 400, 202–209. [Google Scholar] [CrossRef]
- Oberg, K.C. Review of the molecular development of the thumb: Digit primera. Clin. Orthop. Relat. Res. 2014, 472, 1101–1105. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zuniga, A.; Haramis, A.P.; McMahon, A.P.; Zeller, R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature 1999, 401, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Morales-Piga, A.; Bachiller-Corral, J.; Trujillo-Tiebas, M.J.; Villaverde-Hueso, A.; Gamir-Gamir, M.L.; Alonso-Ferreira, V.; Vazquez-Diaz, M.; Posada de la Paz, M.; Ayuso-Garcia, C. Fibrodysplasia ossificans progressiva in Spain: Epidemiological, clinical, and genetic aspects. Bone 2012, 51, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Al Mukaddam, M.; Pignolo, R.J. Acute unilateral hip pain in fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Kitoh, H.; Haga, N.; Nakashima, Y.; Kamizono, J.; Katagiri, T.; Susami, T.; Matsushita, M.; Ishiguro, N. Radiographic characteristics of the hand and cervical spine in fibrodysplasia ossificans progressiva. Intractable Rare Dis. Res. 2014, 3, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.A.; Kaplan, F.S.; Tracy, M.R.; O’Brien, M.L.; Dormans, J.P.; Shore, E.M.; Harland, R.M.; Kusumi, K. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome: Clues from the BMP signaling pathway. Spine 2005, 30, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.R.; Pinnola, G.C.; Ferreira, D.R.A.; Beraldo, P.S.S.; Coelho, C.V.C.; Farage, L.; Takata, R.I.; Speck-Martins, C.E. Mandibular hypoplasia in fibrodysplasia ossificans progressiva causing obstructive sleep apnoea with pulmonary hypertension. Clin. Dysmorphol. 2010, 19, 69–72. [Google Scholar] [CrossRef]
- Carvalho, D.R.; Farage, L.; Martins, B.J.; Speck-Martins, C.E. Craniofacial findings in fibrodysplasia ossificans progressiva: Computerized tomography evaluation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2011, 111, 499–502. [Google Scholar] [CrossRef]
- Carvalho, D.R.; Farage, L.; Speck-Martins, C.E. The signature of craniofacial deformation in fibrodysplasia ossificans progressiva. Am. J. Med. Genet. A 2012, 158A, 2977–2978; author reply 2979–2980. [Google Scholar] [CrossRef]
- Morales-Piga, A.; Bachiller-Corral, J.; Gonzalez-Herranz, P.; Medrano-SanIldelfonso, M.; Olmedo-Garzon, J.; Sanchez-Duffhues, G. Osteochondromas in fibrodysplasia ossificans progressiva: A widespread trait with a streaking but overlooked appearance when arising at femoral bone end. Rheumatol. Int. 2015, 35, 1759–1767. [Google Scholar] [CrossRef]
- Cremin, B.; Connor, J.M.; Beighton, P. The radiological spectrum of fibrodysplasia ossificans progressiva. Clin. Radiol. 1982, 33, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Durbin-Johnson, B.P.; Rocke, D.M.; Kaplan, F.S. Joint-specific risk of impaired function in fibrodysplasia ossificans progressiva (FOP). Bone 2018, 109, 124–133. [Google Scholar] [CrossRef]
- Towler, O.W.; Shore, E.M.; Kaplan, F.S. Skeletal malformations and developmental arthropathy in individuals who have fibrodysplasia ossificans progressiva. Bone 2020, 130, 115116. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Baujat, G.; Brown, M.A.; De Cunto, C.; Hsiao, E.C.; Keen, R.; Al Mukaddam, M.; Le Quan Sang, K.H.; Wilson, A.; Marino, R.; et al. The natural history of fibrodysplasia ossificans progressiva: A prospective, global 36-month study. Genet. Med. 2022, 24, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Toda Nakamura, M.; Hallett, S.A.; Ueharu, H.; Zhang, H.; Kelley, K.; Fukuda, T.; Komatsu, Y.; Mishina, Y. Generation of a new mouse line with conditionally activated signaling through the BMP receptor, ACVR1: A tool to characterize pleiotropic roles of BMP functions. Genesis 2021, 59, e23419. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, L.; Pan, H.; Ueharu, H.; Toda, M.; Yang, Q.; Hallett, S.A.; Olson, L.E.; Mishina, Y. A BMP-controlled metabolic/epigenetic signaling cascade directs midfacial morphogenesis. J. Clin. Investig. 2024, 134, 165787. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Zasloff, M.A.; Kitterman, J.A.; Shore, E.M.; Hong, C.C.; Rocke, D.M. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J. Bone Jt. Surg. Am. 2010, 92, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Storm, E.E.; Kingsley, D.M. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development 1996, 122, 3969–3979. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, M.; Koyama, E.; Shibukawa, Y.; Wu, C.; Tamamura, Y.; Enomoto-Iwamoto, M.; Iwamoto, M. Cellular and molecular mechanisms of synovial joint and articular cartilage formation. Ann. N. Y. Acad. Sci. 2006, 1068, 74–86. [Google Scholar] [CrossRef]
- Koyama, E.; Shibukawa, Y.; Nagayama, M.; Sugito, H.; Young, B.; Yuasa, T.; Okabe, T.; Ochiai, T.; Kamiya, N.; Rountree, R.B.; et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev. Biol. 2008, 316, 62–73. [Google Scholar] [CrossRef]
- Shwartz, Y.; Viukov, S.; Krief, S.; Zelzer, E. Joint Development Involves a Continuous Influx of Gdf5-Positive Cells. Cell Rep. 2016, 15, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Day, T.F.; Jiang, X.; Garrett-Beal, L.; Topol, L.; Yang, Y. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004, 18, 2404–2417. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Wieser, R.; Massague, J.; Niswander, L. Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev. 1997, 11, 2191–2203. [Google Scholar] [CrossRef] [PubMed]
- Lorda-Diez, C.I.; Duarte-Olivenza, C.; Hurle, J.M.; Montero, J.A. Transforming growth factor beta signaling: The master sculptor of fingers. Dev. Dyn. 2022, 251, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Storm, E.E.; Kingsley, D.M. GDF5 coordinates bone and joint formation during digit development. Dev. Biol. 1999, 209, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Towler, O.W.; Shore, E.M.; Xu, M.; Bamford, A.; Anderson, I.; Pignolo, R.J.; Kaplan, F.S. The congenital great toe malformation of fibrodysplasia ossificans progressiva?—A close call. Eur. J. Med. Genet. 2017, 60, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Shirodkar, D.; Smithson, S.F.; Keen, R.; Lester, T.; Banos-Pinero, B.; Burren, C.P. Congenital hallux valgus occurs in Fibrodysplasia Ossificans Progressiva and BMPR1B-associated dysplasia: An important distinction. BMC Med. Genom. 2024, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Baur, S.T.; Mai, J.J.; Dymecki, S.M. Combinatorial signaling through BMP receptor IB and GDF5: Shaping of the distal mouse limb and the genetics of distal limb diversity. Development 2000, 127, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Lyons, K.M.; Rosen, V. BMPs, TGFbeta, and border security at the interzone. Curr. Top. Dev. Biol. 2019, 133, 153–170. [Google Scholar] [CrossRef]
- Lorda-Diez, C.I.; Montero, J.A.; Garcia-Porrero, J.A.; Hurle, J.M. Divergent differentiation of skeletal progenitors into cartilage and tendon: Lessons from the embryonic limb. ACS Chem. Biol. 2014, 9, 72–79. [Google Scholar] [CrossRef]
- Decker, R.S. Articular cartilage and joint development from embryogenesis to adulthood. Semin. Cell Dev. Biol. 2017, 62, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Rountree, R.B.; Schoor, M.; Chen, H.; Marks, M.E.; Harley, V.; Mishina, Y.; Kingsley, D.M. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004, 2, e355. [Google Scholar] [CrossRef] [PubMed]
- Garcia de Vinuesa, A.; Sanchez-Duffhues, G.; Blaney-Davidson, E.; van Caam, A.; Lodder, K.; Ramos, Y.; Kloppenburg, M.; Meulenbelt, I.; van der Kraan, P.; Goumans, M.J.; et al. Cripto favors chondrocyte hypertrophy via TGF-beta SMAD1/5 signaling during development of osteoarthritis. J. Pathol. 2021, 255, 330–342. [Google Scholar] [CrossRef]
- Liu, W.; Feng, M.; Jayasuriya, C.T.; Peng, H.; Zhang, L.; Guan, Y.; Froehlich, J.A.; Terek, R.M.; Chen, Q. Human osteoarthritis cartilage-derived stromal cells activate joint degeneration through TGF-beta lateral signaling. FASEB J. 2020, 34, 16552–16566. [Google Scholar] [CrossRef] [PubMed]
- Di, J.; Chen, Z.; Wang, Z.; He, T.; Wu, D.; Weng, C.; Deng, J.; Mai, L.; Wang, K.; He, L.; et al. Cartilage tissue from sites of weight bearing in patients with osteoarthritis exhibits a differential phenotype with distinct chondrocytes subests. RMD Open 2023, 9, e003255. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Yu, X.; Hsiao, E.C. Cardiopulmonary and Neurologic Dysfunctions in Fibrodysplasia Ossificans Progressiva. Biomedicines 2021, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Daszczuk, P.; Mazurek, P.; Pieczonka, T.D.; Olczak, A.; Boryn, L.M.; Kobielak, K. An Intrinsic Oscillation of Gene Networks Inside Hair Follicle Stem Cells: An Additional Layer That Can Modulate Hair Stem Cell Activities. Front. Cell Dev. Biol. 2020, 8, 595178. [Google Scholar] [CrossRef] [PubMed]
- Kobielak, K.; Stokes, N.; de la Cruz, J.; Polak, L.; Fuchs, E. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 10063–10068. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, M.; Agarwal, S.; Ranganathan, K.; Loder, S.; Cholok, D.; Fireman, D.; Li, J.; Li, S.; Zhao, B.; Mishina, Y.; et al. Hair follicle specific ACVR1/ALK2 critically affects skin morphogenesis and attenuates wound healing. Wound Repair Regen. 2017, 25, 521–525. [Google Scholar] [CrossRef]
- Pulawska-Czub, A.; Pieczonka, T.D.; Mazurek, P.; Kobielak, K. The Potential of Nail Mini-Organ Stem Cells in Skin, Nail and Digit Tips Regeneration. Int. J. Mol. Sci. 2021, 22, 2864. [Google Scholar] [CrossRef]
- Basch, M.L.; Brown, R.M., 2nd; Jen, H.I.; Groves, A.K. Where hearing starts: The development of the mammalian cochlea. J. Anat. 2016, 228, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.M.; Evans, C.C.; Evans, D.A. Cardiopulmonary function in fibrodysplasia ossificans progressiva. Thorax 1981, 36, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Kou, S.; De Cunto, C.; Baujat, G.; Wentworth, K.L.; Grogan, D.R.; Brown, M.A.; Di Rocco, M.; Keen, R.; Al Mukaddam, M.; le Quan Sang, K.H.; et al. Patients with ACVR1(R206H) mutations have an increased prevalence of cardiac conduction abnormalities on electrocardiogram in a natural history study of Fibrodysplasia Ossificans Progressiva. Orphanet J. Rare Dis. 2020, 15, 193. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.S.; Rajderkar, S.; Lane, J.; Mishina, Y.; Kaartinen, V. AcvR1-mediated BMP signaling in second heart field is required for arterial pole development: Implications for myocardial differentiation and regional identity. Dev. Biol. 2014, 390, 191–207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaartinen, V.; Dudas, M.; Nagy, A.; Sridurongrit, S.; Lu, M.M.; Epstein, J.A. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development 2004, 131, 3481–3490. [Google Scholar] [CrossRef] [PubMed]
- Kitterman, J.A.; Strober, J.B.; Kan, L.; Rocke, D.M.; Cali, A.; Peeper, J.; Snow, J.; Delai, P.L.; Morhart, R.; Pignolo, R.J.; et al. Neurological symptoms in individuals with fibrodysplasia ossificans progressiva. J. Neurol. 2012, 259, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Kitterman, J.A.; Procissi, D.; Chakkalakal, S.; Peng, C.Y.; McGuire, T.L.; Goldsby, R.E.; Pignolo, R.J.; Shore, E.M.; Kaplan, F.S.; et al. CNS demyelination in fibrodysplasia ossificans progressiva. J. Neurol. 2012, 259, 2644–2655. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.C.; Di Rocco, M.; Cali, A.; Zasloff, M.; Al Mukaddam, M.; Pignolo, R.J.; Grunwald, Z.; Netelenbos, C.; Keen, R.; Baujat, G.; et al. Special considerations for clinical trials in fibrodysplasia ossificans progressiva (FOP). Br. J. Clin. Pharmacol. 2019, 85, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Al Mukaddam, M.; Pignolo, R.J. A cumulative analogue joint involvement scale (CAJIS) for fibrodysplasia ossificans progressiva (FOP). Bone 2017, 101, 123–128. [Google Scholar] [CrossRef]
- Pacifici, M. Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial. Bone 2018, 109, 267–275. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Nicholas, S.E.; Stoessel, S.J.; Devarakonda, P.M.; Schneider, M.J.; Yamamoto, M.; Goldhamer, D.J. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. eLife 2018, 7, 40814. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Hsiao, E.C.; Al Mukaddam, M.; Baujat, G.; Berglund, S.K.; Brown, M.A.; Cheung, A.M.; De Cunto, C.; Delai, P.; Haga, N.; et al. Reduction of New Heterotopic Ossification (HO) in the Open-Label, Phase 3 MOVE Trial of Palovarotene for Fibrodysplasia Ossificans Progressiva (FOP). J. Bone Miner. Res. 2023, 38, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; Zhong, Y.; He, T.; Chen, S.; Xiao, G. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Forleo-Neto, E.; Pignolo, R.J.; Keen, R.; Orcel, P.; Funck-Brentano, T.; Roux, C.; Kolta, S.; Madeo, A.; Bubbear, J.S.; et al. Garetosmab in fibrodysplasia ossificans progressiva: A randomized, double-blind, placebo-controlled phase 2 trial. Nat. Med. 2023, 29, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Lounev, V.; Groppe, J.C.; Brewer, N.; Wentworth, K.L.; Smith, V.; Xu, M.; Schomburg, L.; Bhargava, P.; Al Mukaddam, M.; Hsiao, E.C.; et al. Matrix metalloproteinase-9 deficiency confers resilience in fibrodysplasia ossificans progressiva in a man and mice. J. Bone Miner. Res. 2024, 39, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Yang, Y. Actionable disease insights from bedside-to-bench investigation in fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2024, 39, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Kim, J.M.; Xie, J.; Chaugule, S.; Lin, C.; Ma, H.; Hsiao, E.; Hong, J.; Chun, H.; Shore, E.M.; et al. Suppression of heterotopic ossification in fibrodysplasia ossificans progressiva using AAV gene delivery. Nat. Commun. 2022, 13, 6175. [Google Scholar] [CrossRef]
- Yang, Y.S.; Lin, C.; Ma, H.; Xie, J.; Kaplan, F.S.; Gao, G.; Shim, J.H. AAV-Mediated Targeting of the Activin A-ACVR1(R206H) Signaling in Fibrodysplasia Ossificans Progressiva. Biomolecules 2023, 13, 1364. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).