


Central Actions of Leptin Induce an Atrophic Pattern and Improves Heart Function in Lean Normoleptinemic Rats via PPARβ/δ Activation

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Intracerebroventricular Leptin Administration
2.3. Pharmacological Administration of the PPARβ/δ Antagonist GSK0660
2.4. Biochemical Assessment
2.5. Quantitative Transcription Analysis with Real-Time Polymerase Chain Reaction (qRT-PCR)
2.6. Western Blot Analysis
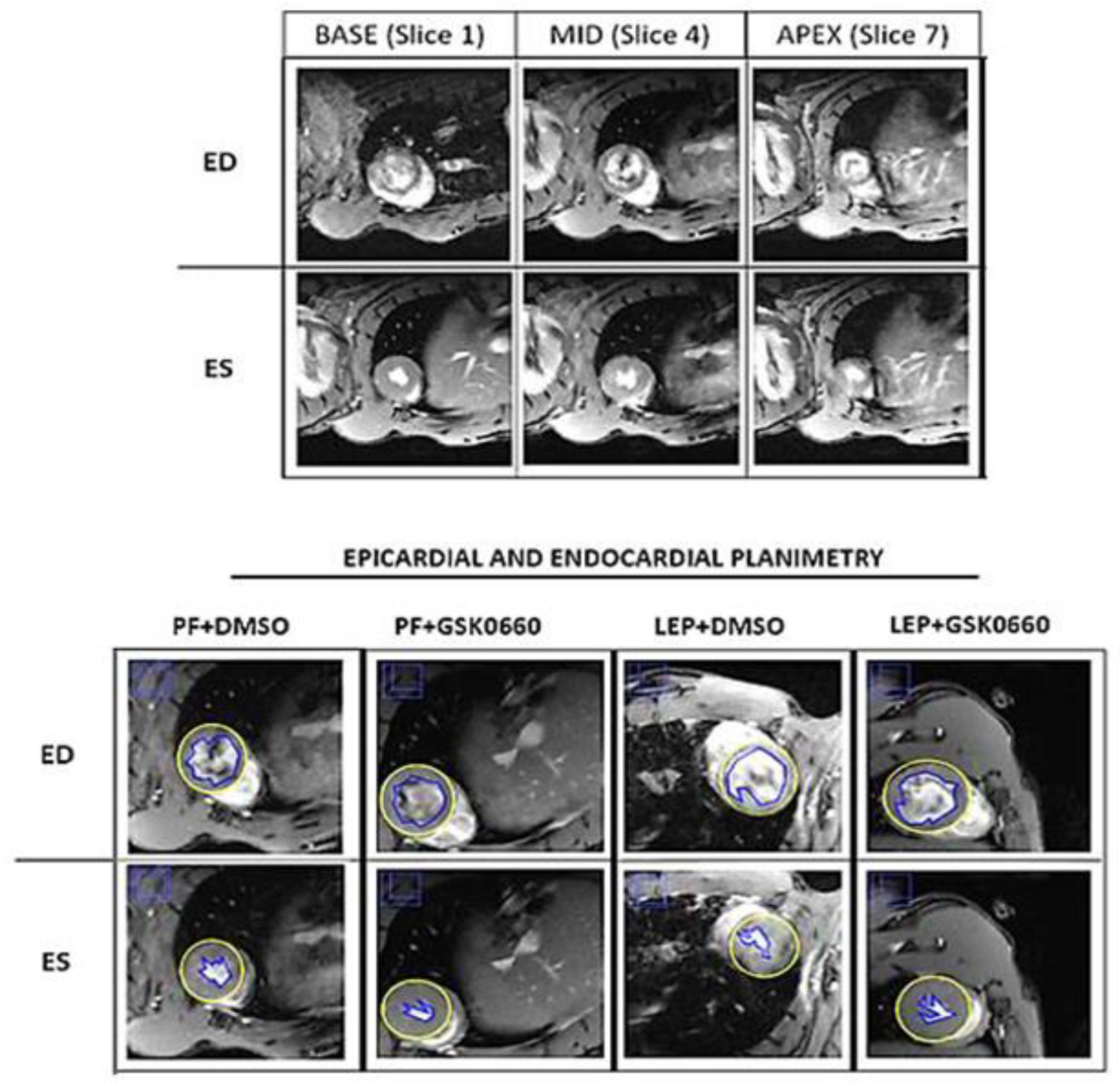
2.7. Cardiac Magnetic Resonance Imaging and Analysis
2.8. Statistical Analysis
3. Results and Discussion
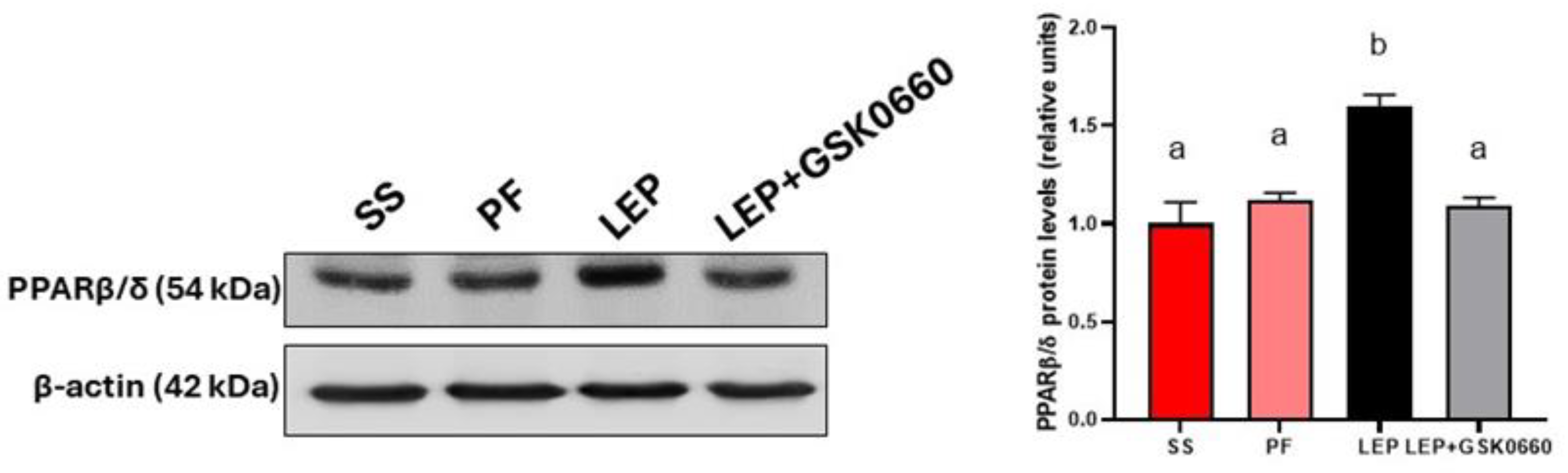
3.1. Validation of Central Leptin Infusion and Pharmacological Inhibition of PPARβ/δ Activity
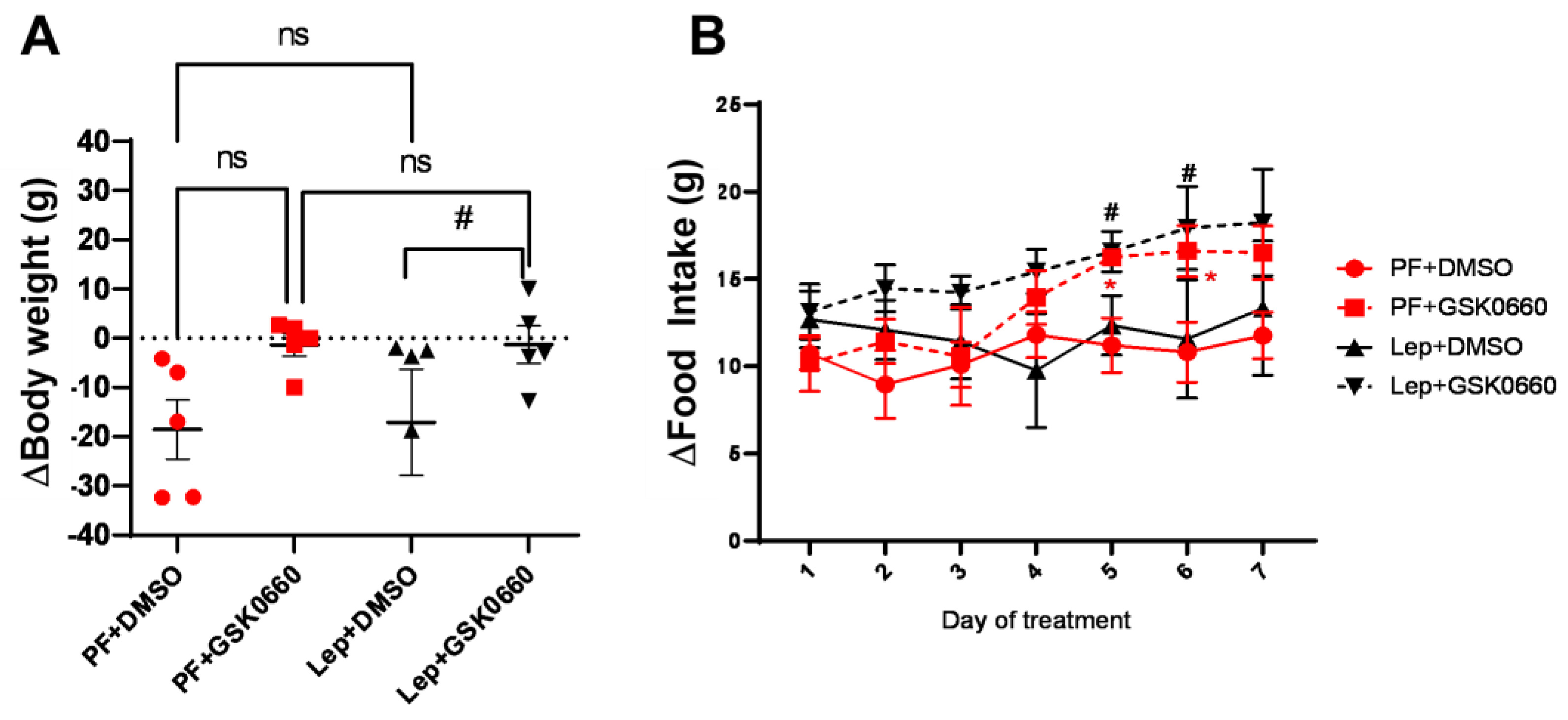
3.2. Effect of Leptin-GSK0660 Co-Treatment on Biological Characteristics of the Rats
3.3. Effect of Leptin-GSK0660 Co-Treatment on Cardiac Electrical Properties
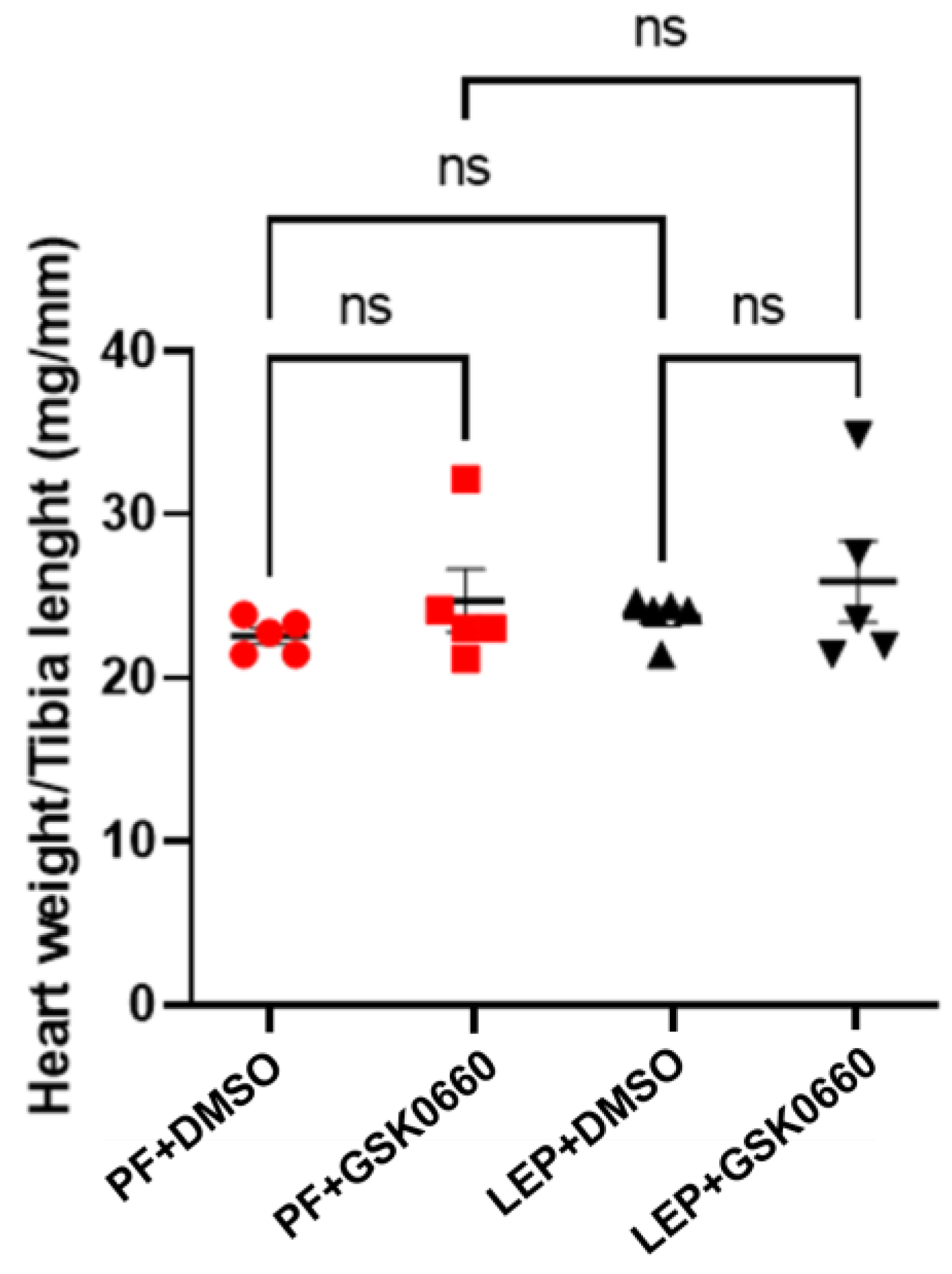
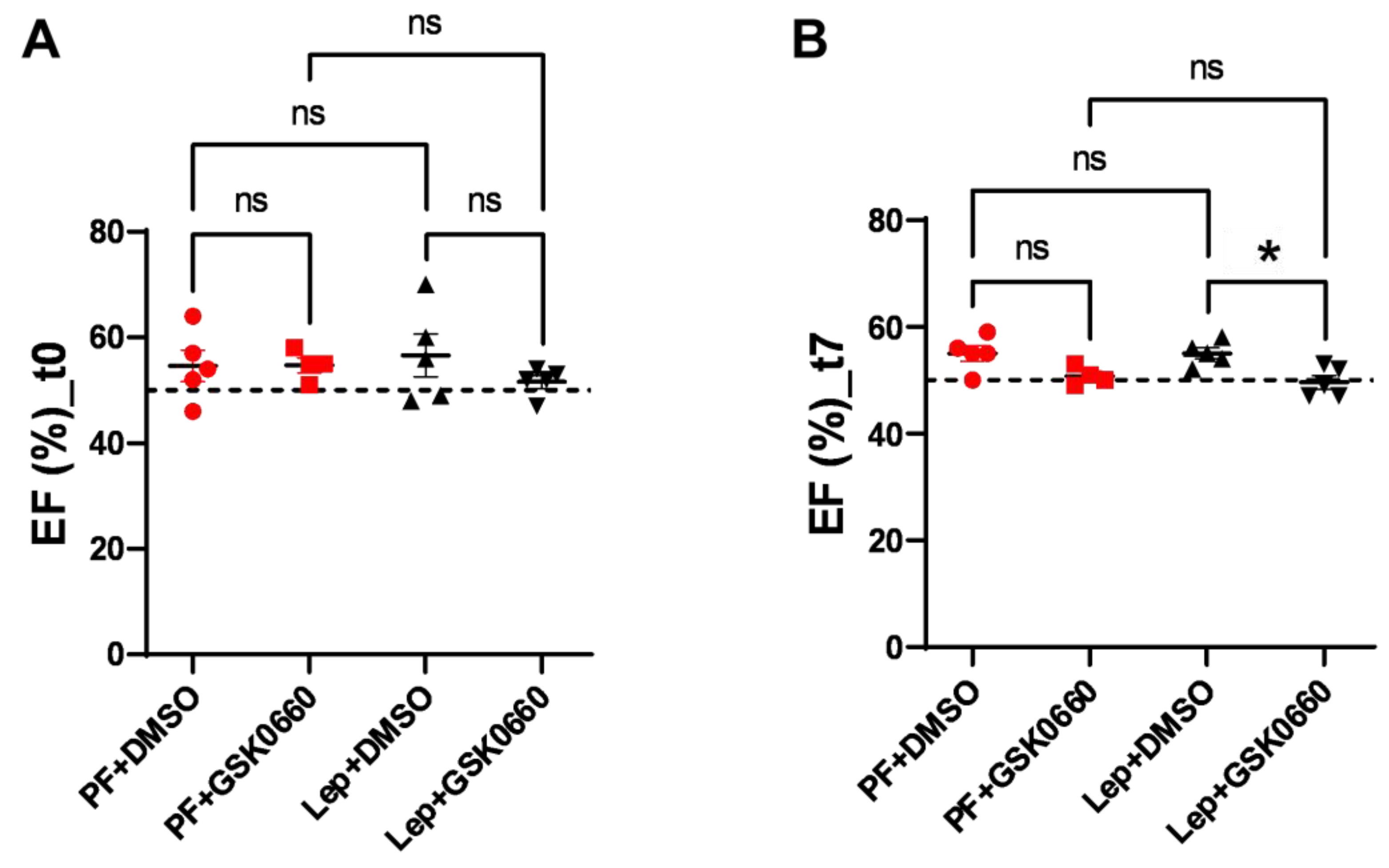
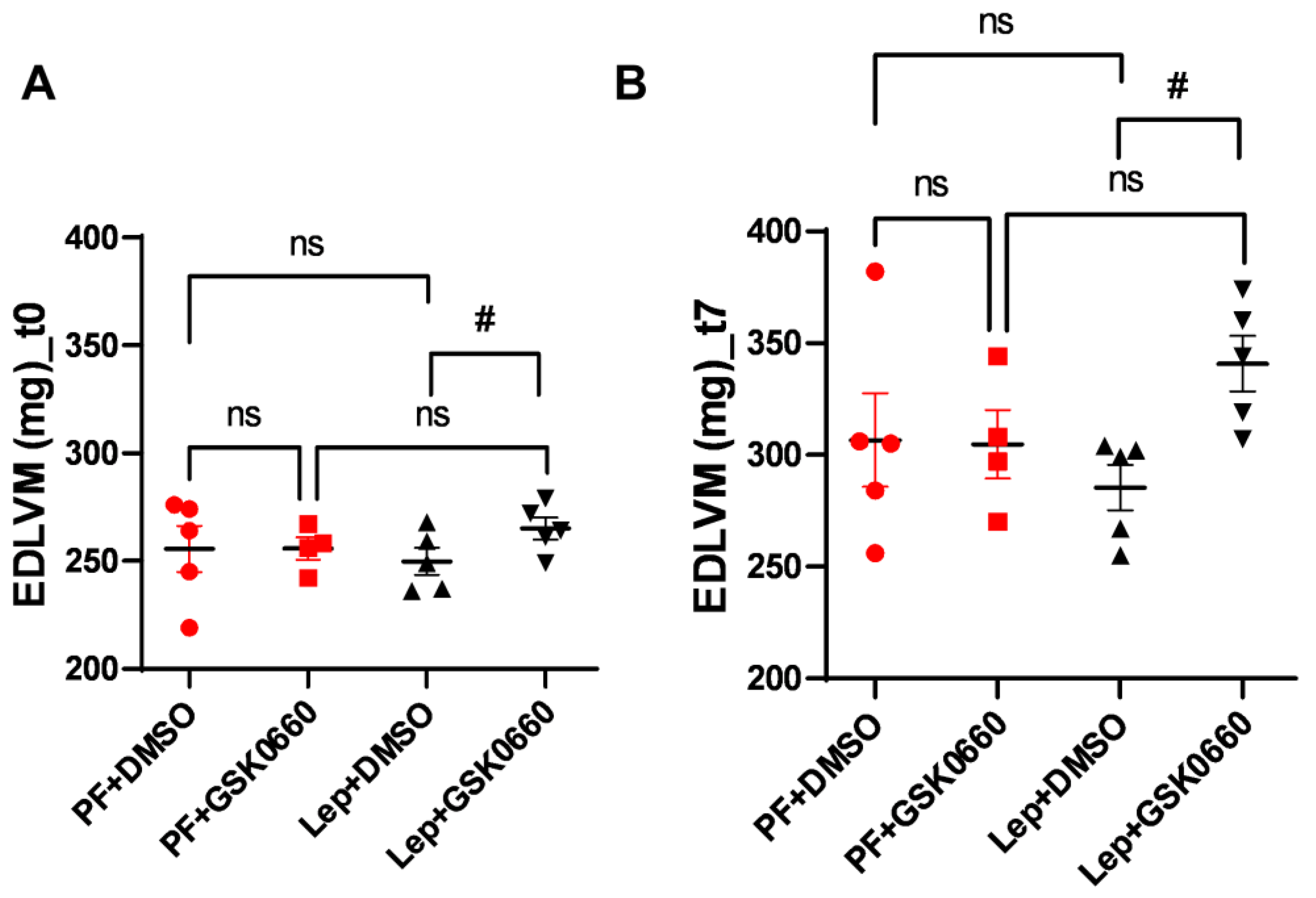
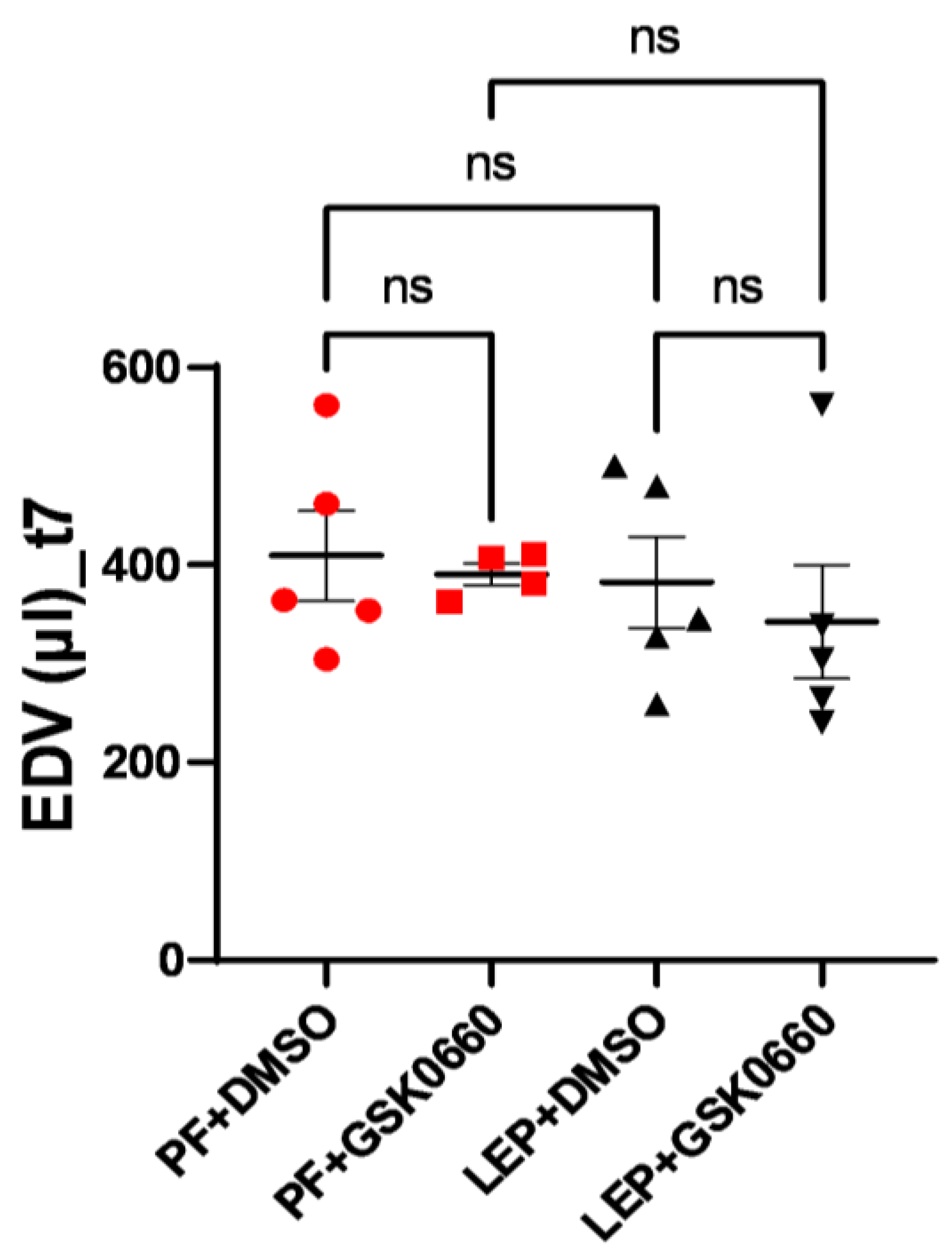
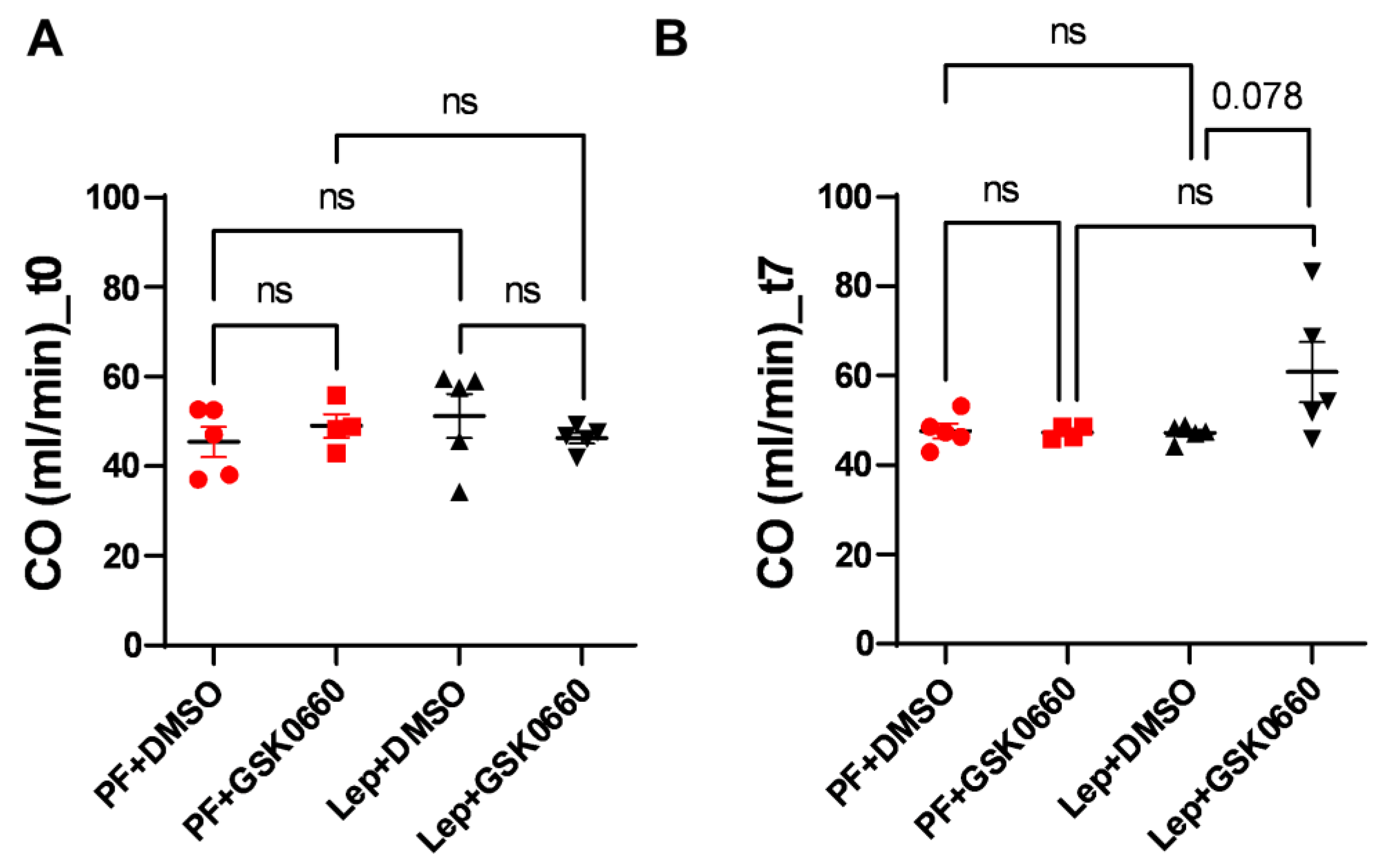
3.4. Effect of Leptin-GSK0660 Co-Treatment on Cardiac Remodeling and Function
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baskin, K.K.; Taegtmeyer, H. Taking Pressure off the Heart: The Ins and Outs of Atrophic Remodelling. Cardiovasc. Res. 2011, 90, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Sudi, S.B.; Tanaka, T.; Oda, S.; Nishiyama, K.; Nishimura, A.; Sunggip, C.; Mangmool, S.; Numaga-Tomita, T.; Nishida, M. TRPC3-Nox2 Axis Mediates Nutritional Deficiency-Induced Cardiomyocyte Atrophy. Sci. Rep. 2019, 9, 9785. [Google Scholar] [CrossRef] [PubMed]
- Bacova, B.S.; Andelova, K.; Sykora, M.; Egan Benova, T.; Barancik, M.; Kurahara, L.H.; Tribulova, N. Does Myocardial Atrophy Represent Anti-Arrhythmic Phenotype? Biomedicines 2022, 10, 2819. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M.; Purdham, D.M.; Rajapurohitam, V.; Zeidan, A. Leptin as a Cardiac Hypertrophic Factor: A Potential Target for Therapeutics. Trends Cardiovasc. Med. 2007, 17, 206–211. [Google Scholar] [CrossRef]
- Hall, M.E.; Harmancey, R.; Stec, D.E. Lean Heart: Role of Leptin in Cardiac Hypertrophy and Metabolism. World J. Cardiol. 2015, 7, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Me, H.; Mw, M.; Je, H.; De, S. Rescue of Cardiac Leptin Receptors in Db/Db Mice Prevents Myocardial Triglyceride Accumulation. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E316–E325. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of Leptin Signaling Contributes to Cardiac Hypertrophy Independently of Body Weight in Mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef]
- Gava, F.N.; da Silva, A.A.; Dai, X.; Harmancey, R.; Ashraf, S.; Omoto, A.C.M.; Salgado, M.C.; Moak, S.P.; Li, X.; Hall, J.E.; et al. Restoration of Cardiac Function After Myocardial Infarction by Long-Term Activation of the CNS Leptin-Melanocortin System. JACC Basic Transl. Sci. 2021, 6, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Suzuki, T.; Wang, W.; de Shazo, M.; Hall, J.E.; Winniford, M.D.; Kullo, I.J.; Mosley, T.H.; Butler, K.R.; Hall, M.E. Higher Plasma Leptin Levels Are Associated with Reduced Left Ventricular Mass and Left Ventricular Diastolic Stiffness in Black Women: Insights from the Genetic Epidemiology Network of Arteriopathy (GENOA) Study. Hypertens. Res. 2018, 41, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Rubio, B.; Mora, C.; Pintado, C.; Mazuecos, L.; Fernández, A.; López, V.; Andrés, A.; Gallardo, N. The Nutrient Sensing Pathways FoxO1/3 and mTOR in the Heart Are Coordinately Regulated by Central Leptin through PPARβ/δ. Implications in Cardiac Remodeling. Metabolism 2020, 115, 154453. [Google Scholar] [CrossRef] [PubMed]
- Mazuecos, L.; Pintado, C.; Rubio, B.; Guisantes-Batán, E.; Andrés, A.; Gallardo, N. Leptin, Acting at Central Level, Increases FGF21 Expression in White Adipose Tissue via PPARβ/δ. Int. J. Mol. Sci. 2021, 22, 4624. [Google Scholar] [CrossRef]
- Rodríguez, M.; Pintado, C.; Torrillas-de la Cal, R.; Moltó, E.; Gallardo, N.; Andrés, A.; Arribas, C. Ageing Alters the Lipid Sensing Process in the Hypothalamus of Wistar Rat. Effect of Food Restriction. Nutr. Neurosci. 2022, 25, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; Pintado, C.; Rubio, B.; Mazuecos, L.; López, V.; Fernández, A.; Salamanca, A.; Bárcena, B.; Fernández-Agulló, T.; Arribas, C.; et al. Central Leptin Regulates Heart Lipid Content by Selectively Increasing PPAR β/δ Expression. J. Endocrinol. 2018, 236, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Heiberg, E.; Sjögren, J.; Ugander, M.; Carlsson, M.; Engblom, H.; Arheden, H. Design and Validation of Segment--Freely Available Software for Cardiovascular Image Analysis. BMC Med. Imaging 2010, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Schweins, M.; Gäbel, R.; Raitza, M.; Vasudevan, P.; Lemcke, H.; Joksch, M.; Schildt, A.; Kurth, J.; Lindner, T.; Meinel, F.G.; et al. Multi-Modal Assessment of a Cardiac Stem Cell Therapy Reveals Distinct Modulation of Regional Scar Properties. J. Transl. Med. 2024, 22, 187. [Google Scholar] [CrossRef]
- Lang, C.I.; Vasudevan, P.; Döring, P.; Gäbel, R.; Lemcke, H.; Lindner, T.; Steinhoff, G.; Krause, B.J.; Vollmar, B.; Meinel, F.G.; et al. Expedient Assessment of Post-Infarct Remodeling by Native Cardiac Magnetic Resonance Imaging in Mice. Sci. Rep. 2021, 11, 11625. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.C.; Spurgeon, H.A.; Rakusan, K.; Weisfeldt, M.L.; Lakatta, E.G. Use of Tibial Length to Quantify Cardiac Hypertrophy: Application in the Aging Rat. Am. J. Physiol. 1982, 243, H941–H947. [Google Scholar] [CrossRef] [PubMed]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.-F.; Dohmen, P.M.; Choi, Y.-H.; Wahlers, T.; et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Domain, G.; Chouquet, C.; Réant, P.; Bongard, V.; Vedis, T.; Rollin, A.; Mandel, F.; Delasnerie, H.; Voglimacci-Stephanopoli, Q.; Mondoly, P.; et al. Relationships between Left Ventricular Mass and QRS Duration in Diverse Types of Left Ventricular Hypertrophy. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Okin, P.M.; Roman, M.J.; Devereux, R.B.; Kligfield, P. Gender Differences and the Electrocardiogram in Left Ventricular Hypertrophy. Hypertension 1995, 25, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, I.; Galatou, E.; Panagiotidis, G.-D.; Ravingerová, T.; Lazou, A. Cardioprotective Effects of PPARβ/δ Activation against Ischemia/Reperfusion Injury in Rat Heart Are Associated with ALDH2 Upregulation, Amelioration of Oxidative Stress and Preservation of Mitochondrial Energy Production. Int. J. Mol. Sci. 2021, 22, 6399. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Rodríguez-Calvo, R.; Jové, M.; Michalik, L.; Wahli, W.; Laguna, J.C.; Vázquez-Carrera, M. Peroxisome Proliferator-Activated Receptor Beta/Delta Activation Inhibits Hypertrophy in Neonatal Rat Cardiomyocytes. Cardiovasc. Res. 2005, 65, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Barish, G.D.; Narkar, V.A.; Evans, R.M. PPAR Delta: A Dagger in the Heart of the Metabolic Syndrome. J. Clin. Investig. 2006, 116, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in Skeletal Muscle Physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef] [PubMed]
- DeBoer, M.D.; Gurka, M.J.; Woo, J.G.; Morrison, J.A. Severity of Metabolic Syndrome as a Predictor of Cardiovascular Disease Between Childhood and Adulthood: The Princeton Lipid Research Cohort Study. J. Am. Coll. Cardiol. 2015, 66, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Olson, P.; Evans, R.M. Minireview: Lipid Metabolism, Metabolic Diseases, and Peroxisome Proliferator-Activated Receptors. Endocrinology 2003, 144, 2201–2207. [Google Scholar] [CrossRef]
- Wang, Y.-X. PPARs: Diverse Regulators in Energy Metabolism and Metabolic Diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Huypens, P.; Stewart, L.K.; Gettys, T.W. Implications of Crosstalk between Leptin and Insulin Signaling during the Development of Diet-Induced Obesity. Biochim. Biophys. Acta 2009, 1792, 409–416. [Google Scholar] [CrossRef]
- Sierra-Johnson, J.; Romero-Corral, A.; Lopez-Jimenez, F.; Gami, A.S.; Sert Kuniyoshi, F.H.; Wolk, R.; Somers, V.K. Relation of Increased Leptin Concentrations to History of Myocardial Infarction and Stroke in the United States Population. Am. J. Cardiol. 2007, 100, 234–239. [Google Scholar] [CrossRef]
- Hori, M.; Okamoto, H. Heart Rate as a Target of Treatment of Chronic Heart Failure. J. Cardiol. 2012, 60, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Fraley, M.A.; Birchem, J.A.; Senkottaiyan, N.; Alpert, M.A. Obesity and the Electrocardiogram. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2005, 6, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Wiegerinck, R.F.; Verkerk, A.O.; Belterman, C.N.; van Veen, T.A.B.; Baartscheer, A.; Opthof, T.; Wilders, R.; de Bakker, J.M.T.; Coronel, R. Larger Cell Size in Rabbits with Heart Failure Increases Myocardial Conduction Velocity and QRS Duration. Circulation 2006, 113, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Huang, J.; Hileman, S.; Martin, K.H.; Hull, R.; Davis, M.; Yu, H.-G. Leptin Decreases Heart Rate Associated with Increased Ventricular Repolarization via Its Receptor. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1731–H1739. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for Cardiac Chamber Quantification by Echocardiography in Adults: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. Off. Publ. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef]
- Savarese, G.; Stolfo, D.; Sinagra, G.; Lund, L.H. Heart Failure with Mid-Range or Mildly Reduced Ejection Fraction. Nat. Rev. Cardiol. 2022, 19, 100–116. [Google Scholar] [CrossRef]
- Golla, M.S.G.; Hajouli, S.; Ludhwani, D. Heart Failure and Ejection Fraction. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin Resistance: Metabolic Mechanisms and Consequences in the Heart. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Mitochondrial Dynamics in Heart Disease. Biochim. Biophys. Acta 2013, 1833, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Saheera, S.; Krishnamurthy, P. Cardiovascular Changes Associated with Hypertensive Heart Disease and Aging. Cell Transplant. 2020, 29, 963689720920830. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Izumiya, Y.; Maatz, H.; Razeghi, P.; Shiojima, I.; Sandri, M.; Sato, K.; Zeng, L.; Schiekofer, S.; Pimentel, D.; et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 2005, 280, 20814–20823. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-L.; Sachdev, V.; Burklow, T.R.; Li, W.; Startzell, M.; Auh, S.; Brown, R.J. Leptin Attenuates Cardiac Hypertrophy in Patients With Generalized Lipodystrophy. J. Clin. Endocrinol. Metab. 2021, 106, e4327–e4339. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repetition time | 8 ms |
| Echo time | 2.5 ms |
| Slices thickness | 1.5 mm |
| FOV | 40 × 40 mm |
| Matrix | 256 × 256 |
| Resolution | 0.156 × 0.156 |
| Hypothalamic Gene | SS | PF | LEP |
|---|---|---|---|
| Ob-Rb | 1.0 ± 0.3 a | 2.7 ± 0.2 b | 3.7 ± 0.4 c |
| Crh | 0.9 ± 0.1 a | 0.6 ± 0.1 b | 1.96 ± 0.1 c |
| Trh | 1.1 ± 0.1 a | 0.9 ± 0.1 a | 2.6 ± 0.3 b |
| PF + DMSO | PF + GSK0660 | LEP + DMSO | LEP + GSK0660 | |
|---|---|---|---|---|
| PrWAT (g) | 4.62 ± 0.86 a | 4.86 ± 1 a | 3.04 ± 0.85 a | 3.71 ± 0.68 a |
| eWAT (g) | 4.21 ± 0.36 a | 4.80 ± 0.86 a | 3.10 ± 0.54 a | 3.53 ± 0.40 a |
| BAT (g) | 0.42 ± 0.05 a | 0.40 ± 0.07 a | 0.29 ± 0.02 a | 0.39 ± 0.04 a |
| Serum glucose (mg/dL) | 99 ± 19 a | 105 ± 12 a | 112 ± 12 a | 118 ± 10 a |
| Serum leptin (ng/mL) | 1.07 ± 0.1 a | 1.60 ± 0.5 a | 1.10 ± 0.3 a | 1.14 ± 0.3 a |
| Serum insulin (ng/mL) | 2.49 ± 0.8 a | 2.67 ± 0.6 a | 2.46 ± 0.6 a | 5.19 ± 2.1 b |
| QRS Duration (ms) | ∆QRS (ms) | R Amplitude (mV) | ∆R (mV) | |||
| t0 | t7 | t0 | t7 | |||
| PF+DMSO | 22.7 ± 5.1 | 19.6 ± 5.1 | −3.1 ± 3.2 a | 147 ± 19 | 149 ± 24 | 2.0 ± 15 a |
| PF+GSK0660 | 28.8 ± 5.7 | 26.3 ± 5.0 | −2.5 ± 3.6 a | 103 ± 26 | 95 ± 7 | −8 ± 14 a |
| LEP+DMSO | 29.2 ± 7.1 | 20.4 ± 3.8 * | −8.8 ± 3.6 a | 137 ± 23 | 93 ± 12 * | −44 ± 13 b |
| LEP+GSK0660 | 21.4 ± 3.0 | 17.1 ± 8 | −4.3 ± 3.8 a | 126 ± 12 | 132 ± 27 | 6 ± 13 a |
| QT Duration (ms) | ∆QT (ms) | QTc Duration (ms) | ∆QTc (ms) | |||
| t0 | t7 | t0 | t7 | |||
| PF+DMSO | 71.9 ± 13.4 | 70.9 ± 11.1 | −1.0 ± 7.8 a | 6.2 ± 1.2 | 5.6 ± 0.8 | −0.6 ± 0.6 a |
| PF+GSK0660 | 65.2 ± 7.3 | 55.1 ± 7.3 | −10.1 ± 4.9 a,b | 5.4 ± 0.6 | 4.2 ± 0.5 * | −1.2 ± 0.4 a |
| LEP+DMSO | 79.9 ± 10.5 | 65.5 ± 12.1 | −14.4 ± 7.2 b | 6.9 ± 1.0 | 5.6 ± 1.1 | −1.3 ± 0.7 a |
| LEP+GSK0660 | 55.3 ± 7.2 | 60.3 ± 3.6 | 5.0 ± 3.6 a,c | 4.6 ± 0.6 | 5.1 ± 0.3 | 0.5 ± 0.3 b |
| R-R INTERVAL (ms) | ∆R-R (ms) | Heart Rate (bpm) | ∆HR (bpm) | |||
|---|---|---|---|---|---|---|
| t0 | t7 | t0 | t7 | |||
| PF+DMSO | 136 ± 6 | 159 ± 7 * | 23 ± 4.1 a | 210 ± 22 | 274 ± 30 * | 64 ± 16.64 a |
| PF+GSK0660 | 145 ± 4 | 196 ± 31 * | 51 ± 14.0 b | 205 ± 12 | 275 ± 34 * | 70 ± 16.12 a |
| LEP+DMSO | 135 ± 5 | 139 ± 4 | 4 ± 2.9 c | 231 ± 30 | 219 ± 20 | −12 ± 16.12 b |
| LEP+GSK0660 | 141 ± 4 | 142 ± 6 | 1 ± 3.2 c | 199 ± 4 | 224 ± 14 * | 25 ± 6.51 c |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio, B.; Pintado, C.; Mazuecos, L.; Benito, M.; Andrés, A.; Gallardo, N. Central Actions of Leptin Induce an Atrophic Pattern and Improves Heart Function in Lean Normoleptinemic Rats via PPARβ/δ Activation. Biomolecules 2024, 14, 1028. https://doi.org/10.3390/biom14081028
Rubio B, Pintado C, Mazuecos L, Benito M, Andrés A, Gallardo N. Central Actions of Leptin Induce an Atrophic Pattern and Improves Heart Function in Lean Normoleptinemic Rats via PPARβ/δ Activation. Biomolecules. 2024; 14(8):1028. https://doi.org/10.3390/biom14081028
Chicago/Turabian StyleRubio, Blanca, Cristina Pintado, Lorena Mazuecos, Marina Benito, Antonio Andrés, and Nilda Gallardo. 2024. "Central Actions of Leptin Induce an Atrophic Pattern and Improves Heart Function in Lean Normoleptinemic Rats via PPARβ/δ Activation" Biomolecules 14, no. 8: 1028. https://doi.org/10.3390/biom14081028
APA StyleRubio, B., Pintado, C., Mazuecos, L., Benito, M., Andrés, A., & Gallardo, N. (2024). Central Actions of Leptin Induce an Atrophic Pattern and Improves Heart Function in Lean Normoleptinemic Rats via PPARβ/δ Activation. Biomolecules, 14(8), 1028. https://doi.org/10.3390/biom14081028