

FGF1 Suppresses Allosteric Activation of β3 Integrins by FGF2: A Potential Mechanism of Anti-Inflammatory and Anti-Thrombotic Action of FGF1



Abstract
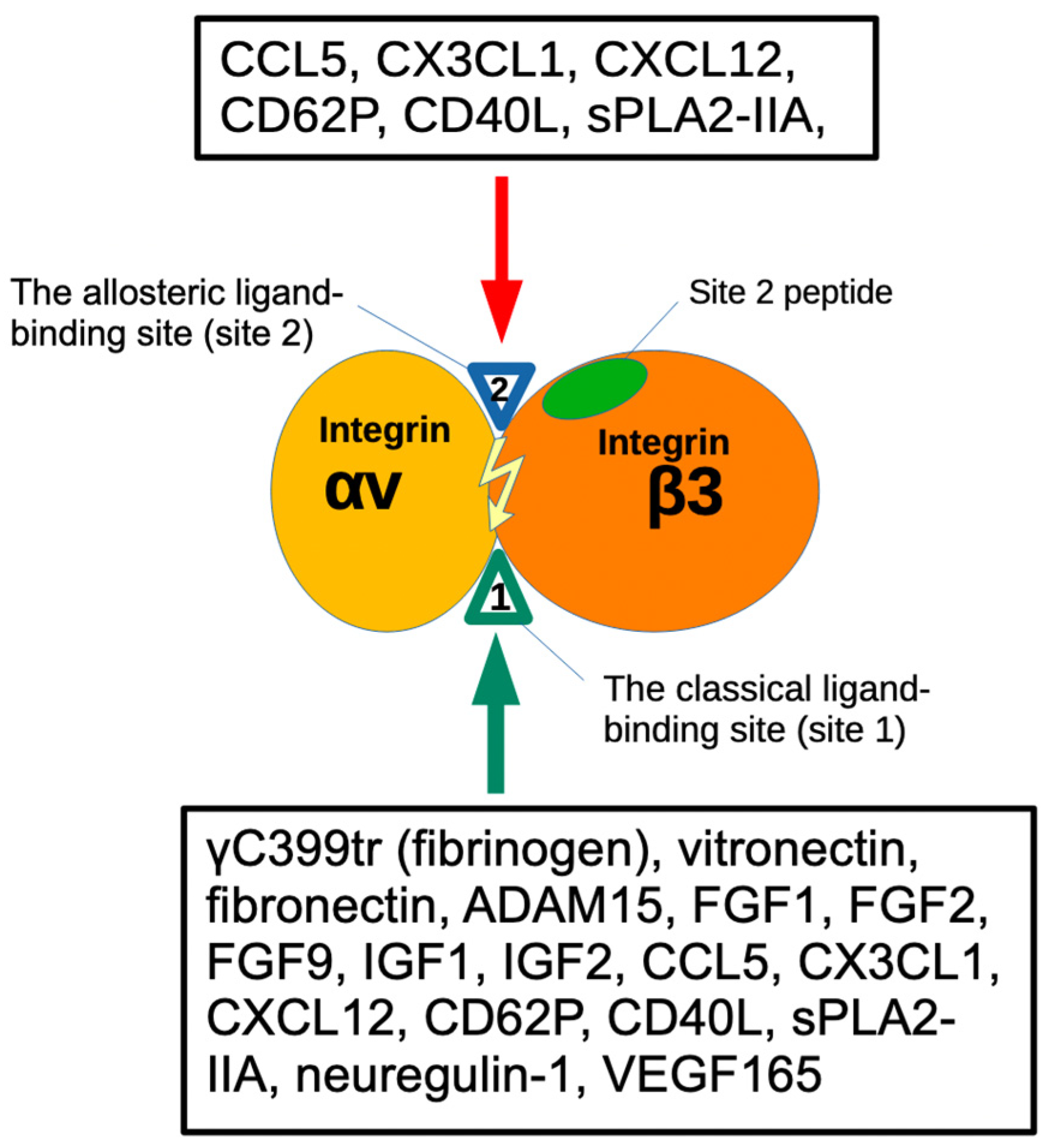
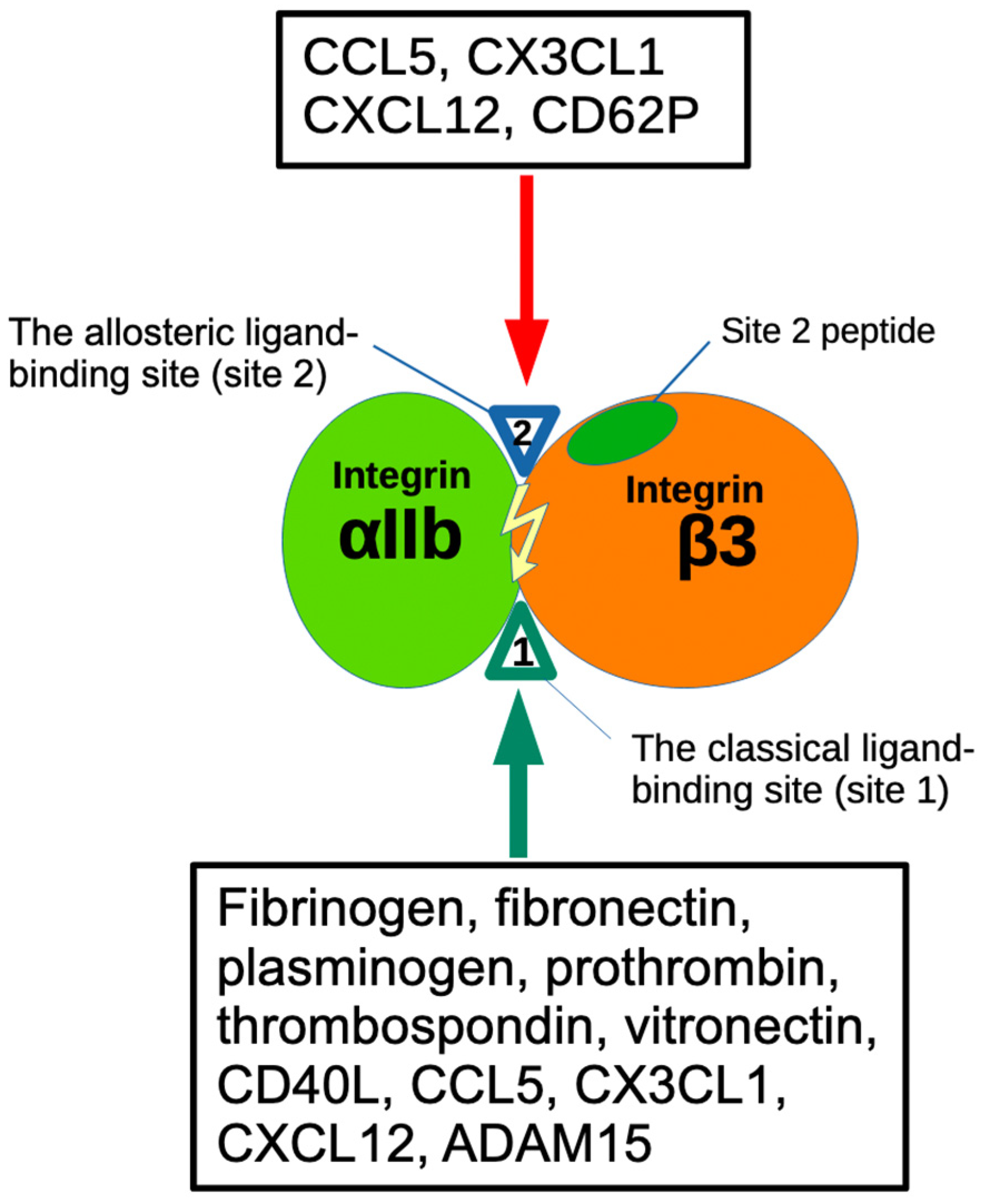
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Activation of Soluble αIIbβ3 and αvβ3 by FGF2
2.3. Activation of Integrin αIIbβ3 on the Cell Surface by FGF2
2.4. Binding of Site 2 Peptide to FGF1 and FGF2
2.5. Docking Simulation
2.6. Statistical Analysis
3. Results
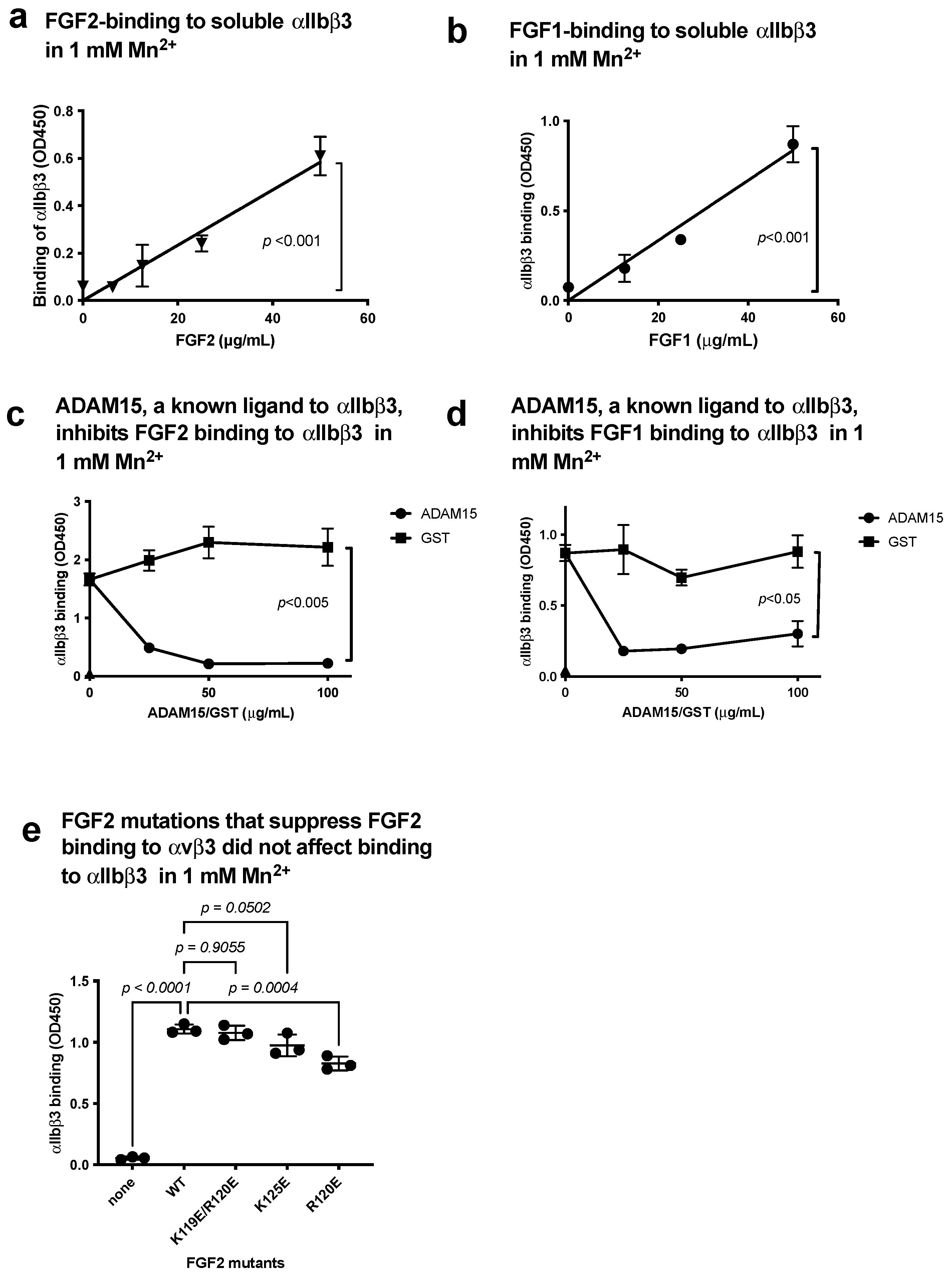
3.1. FGF1 and FGF2 Bind to the Classical Binding Site (Site 1) of αIIbβ3
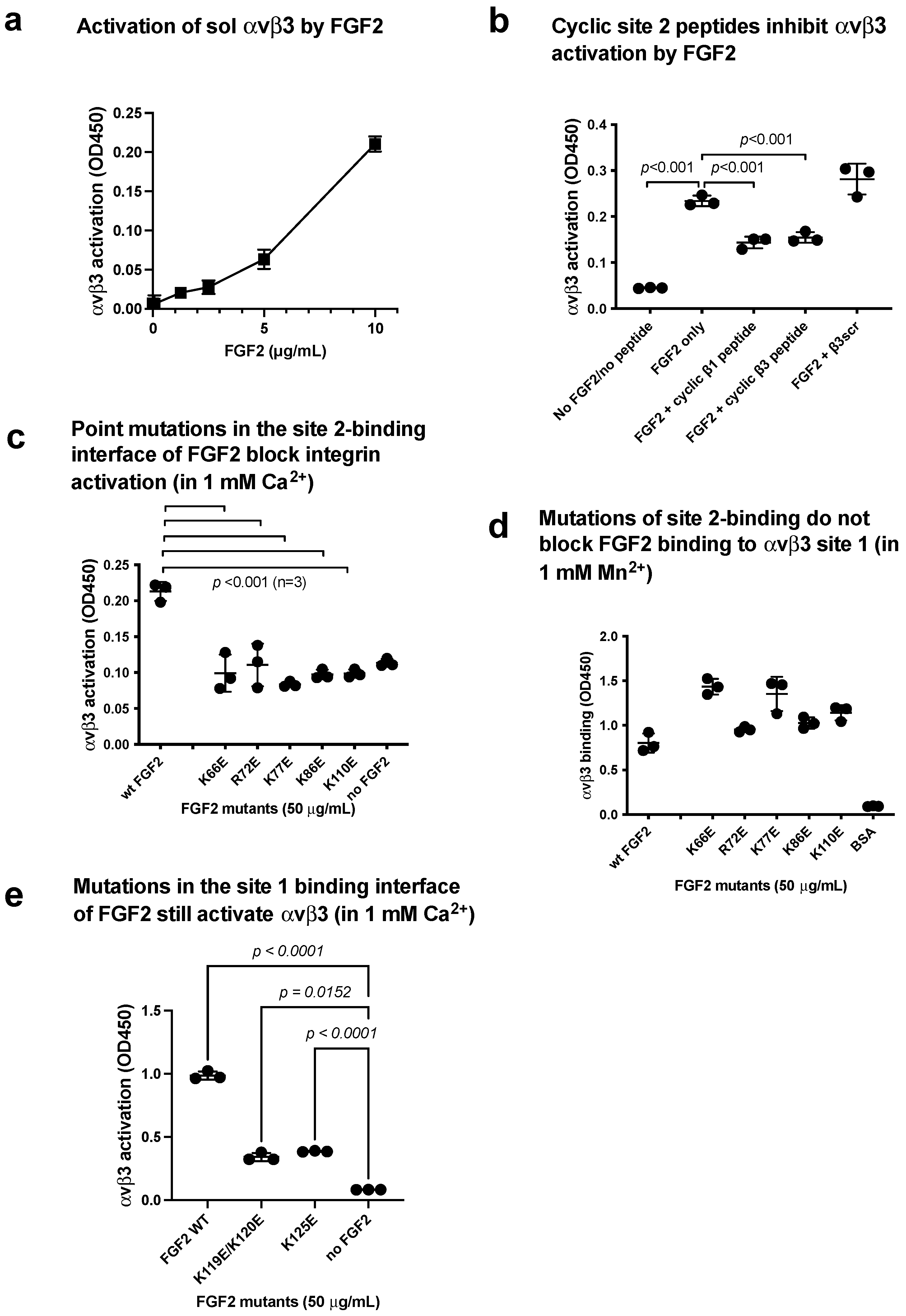
3.2. FGF2 Activates Soluble Integrin αIIbβ3 by Binding to the Allosteric Site (Site 2)
3.3. Point Mutations of the Predicted Site 2-Binding Interface in FGF2 Block FGF2-Mediated Activation of αIIbβ3
3.4. FGF2 Activates Soluble Integrin αvβ3 by Binding to the Allosteric Site (Site 2)
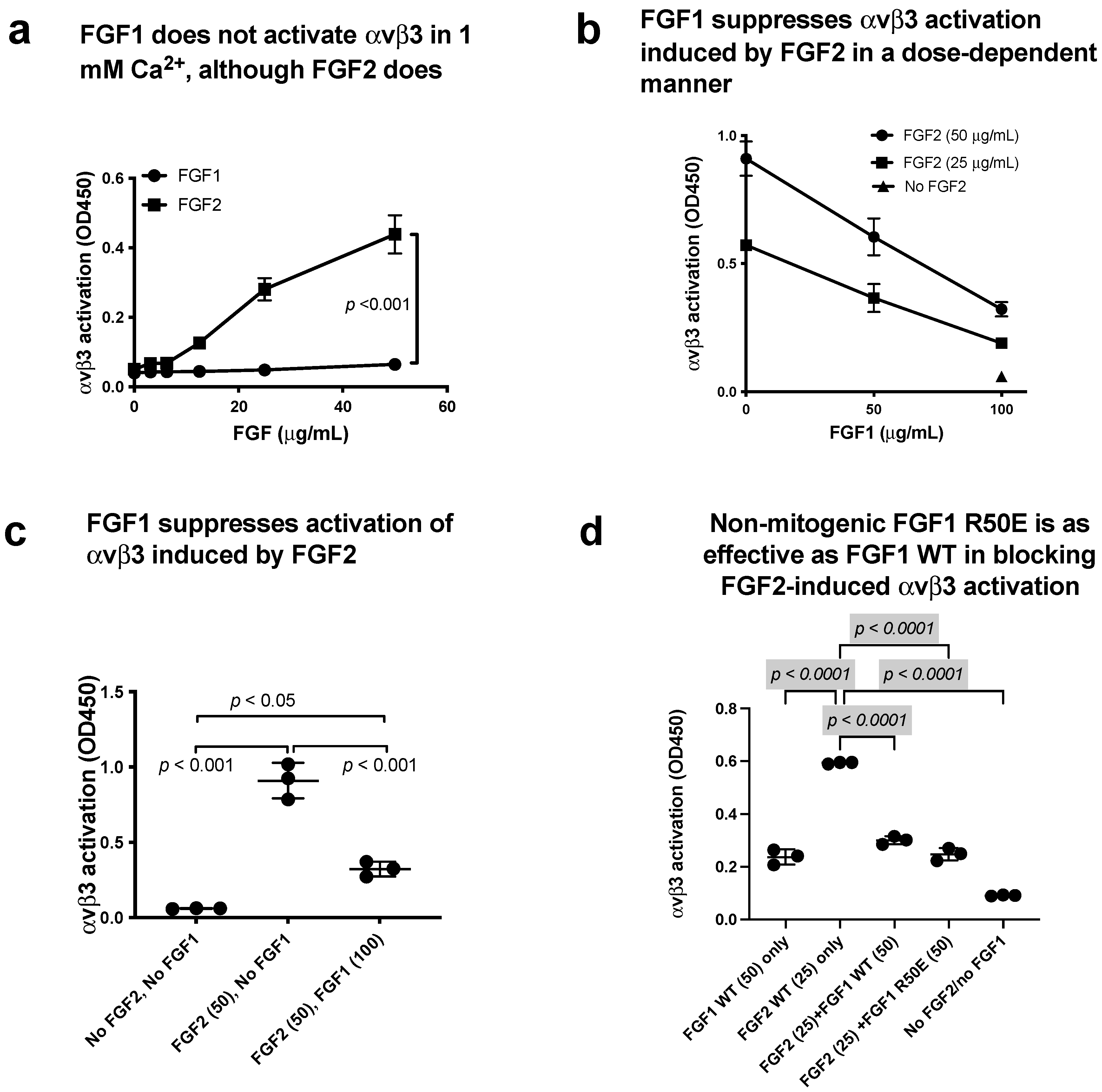
3.5. FGF1 Binds to Site 2 but Does Not Activate β3 Integrins and Suppresses β3 Integrin Activation Induced by FGF2
3.6. Non-Mitogenic FGF1 R50E Mutant Suppressed FGF2-Induced Activation of β3 Integrins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Clark, R.A.; Cheresh, D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 1994, 264, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, D.A.; Stupack, D.G. Regulation of angiogenesis: Apoptotic cues from the ECM. Oncogene 2008, 27, 6285–6298. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Wu, C.-Y.; Yamaji, S.; Saegusa, J.; Shi, B.; Ma, Z.; Kuwabara, Y.; Lam, K.S.; Isseroff, R.R.; Takada, Y.K.; et al. Direct binding of integrin alphavbeta3 to FGF1 plays a role in FGF1 signaling. J. Biol. Chem. 2008, 283, 18066–18075. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Hatori, N.; Kawaguchi, N.; Hamada, Y.; Shih, T.-C.; Wu, C.-Y.; Lam, K.S.; Matsuura, N.; Yamamoto, H.; Takada, Y.K.; et al. The integrin-binding defective FGF2 mutants potently suppress FGF2 signaling and angiogenesis. Biosci. Rep. 2017, 37, BSR20170173. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, S.; Saegusa, J.; Ieguchi, K.; Fujita, M.; Mori, S.; Takada, Y.K.; Takada, Y. A novel fibroblast growth factor-1 (FGF1) mutant that acts as an FGF antagonist. PLoS ONE 2010, 5, e10273. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Tran, V.; Nishikawa, K.; Kaneda, T.; Hamada, Y.; Kawaguchi, N.; Fujita, M.; Takada, Y.K.; Matsuura, N.; Zhao, M.; et al. A dominant-negative FGF1 mutant (the R50E mutant) suppresses tumorigenesis and angiogenesis. PLoS ONE 2013, 8, e57927. [Google Scholar] [CrossRef]
- Cheng, Y.-S.; Taniguchi, Y.; Yunoki, Y.; Masai, S.; Nogi, M.; Doi, H.; Sekiguchi, K.; Nakagawa, M. Simultaneous binding of bFGF to both FGFR and integrin maintains properties of primed human induced pluripotent stem cells. Regen. Ther. 2024, 25, 113–127. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef]
- Ginsberg, M.H. Integrin activation. BMB. Rep. 2014, 47, 655–659. [Google Scholar] [CrossRef]
- Fujita, M.; Takada, Y.K.; Takada, Y. The chemokine fractalkine can activate integrins without CX3CR1 through direct binding to a ligand-binding site distinct from the classical RGD-binding site. PLoS ONE 2014, 9, e96372. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Davari, P.; Takada, Y.K.; Takada, Y. Stromal cell-derived factor-1 (CXCL12) activates integrins by direct binding to an allosteric ligand-binding site (site 2) of integrins without CXCR4. Biochem. J. 2018, 475, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.K.; Fujita, M.; Takada, Y. Pro-Inflammatory Chemokines CCL5, CXCL12, and CX3CL1 Bind to and Activate Platelet Integrin alphaIIbbeta3 in an Allosteric Manner. Cells 2022, 11, 3059. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Zhu, K.; Fujita, C.K.; Zhao, M.; Lam, K.S.; Kurth, M.J.; Takada, Y.K.; Takada, Y. Proinflammatory secreted phospholipase A2 type IIA (sPLA-IIA) induces integrin activation through direct binding to a newly identified binding site (site 2) in integrins alphavbeta3, alpha4beta1, and alpha5beta1. J. Biol. Chem. 2015, 290, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.K.; Shimoda, M.; Maverakis, E.; Felding, B.H.; Cheng, R.H.; Takada, Y. Soluble CD40L activates soluble and cell-surface integrins alphavbeta3, alpha5beta1 and alpha4beta1 by binding to the allosteric ligand-binding site (site 2). J. Biol. Chem. 2021, 296, 100399. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.K.; Shimoda, M.; Takada, Y. The C-type lectin domain of CD62P (P-selectin) functions as an integrin ligand. Life Sci. Alliance 2023, 6, e202201747. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Takada, Y.K.; Takada, Y. Integrins alphavbeta3 and alpha4beta1 act as coreceptors for fractalkine, and the integrin-binding defective mutant of fractalkine is an antagonist of CX3CR1. J. Immunol. 2012, 189, 5809–5819. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.M.; Shil, N.K.; Gc, J.B.; Colburn, Z.T.; Tsai, S.-Y.; Segovia, J.A.; Chang, T.-H.; Bandyopadhyay, S.; Natesan, S.; Jones, J.C.R.; et al. Integrin activation by the lipid molecule 25-hydroxycholesterol induces a proinflammatory response. Nat. Commun. 2019, 10, 1482. [Google Scholar] [CrossRef]
- Zhang, X.-P.; Kamata, T.; Yokoyama, K.; Puzon-McLaughlin, W.; Takada, Y. Specific interaction of the recombinant disintegrin-like domain of MDC-15 (metargidin, ADAM-15) with integrin alphavbeta3. J. Biol. Chem. 1998, 273, 7345–7350. [Google Scholar] [CrossRef]
- Chang, C.-C.; Takada, Y.K.; Cheng, C.-W.; Maekawa, Y.; Mori, S.; Takada, Y. FGF9, a Potent Mitogen, Is a New Ligand for Integrin alphavbeta3, and the FGF9 Mutant Defective in Integrin Binding Acts as an Antagonist. Cells 2024, 13, 307. [Google Scholar] [CrossRef]
- Takada, Y.K.; Yu, J.; Ye, X.; Wu, C.-Y.; Felding, B.H.; Fujita, M.; Takada, Y. The heparin-binding domain of VEGF165 directly binds to integrin alphavbeta3 and VEGFR2/KDR D1: A potential mechanism of negative regulation of VEGF165 signaling by alphavbeta3. Front. Cell Dev. Biol. 2024, 12, 1347616. [Google Scholar] [CrossRef]
- Saegusa, J.; Yamaji, S.; Ieguchi, K.; Wu, C.-Y.; Lam, K.S.; Liu, F.-T.; Takada, Y.K.; Takada, Y. The direct binding of insulin-like growth factor-1 (IGF-1) to integrin alphavbeta3 is involved in IGF-1 signaling. J. Biol. Chem. 2009, 284, 24106–24114. [Google Scholar] [CrossRef]
- Prieto, D.M.C.; Cheng, Y.; Chang, C.-C.; Yu, J.; Takada, Y.K.; Takada, Y. Direct integrin binding to insulin-like growth factor-2 through the C-domain is required for insulin-like growth factor receptor type 1 (IGF1R) signaling. PLoS ONE 2017, 12, e0184285. [Google Scholar] [CrossRef]
- Takada, Y.K.; Shimoda, M.; Takada, Y. CD40L Activates Platelet Integrin alphaIIbbeta3 by Binding to the Allosteric Site (Site 2) in a KGD-Independent Manner and HIGM1 Mutations Are Clustered in the Integrin-Binding Sites of CD40L. Cells 2023, 12, 1977. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.K.; Yu, J.; Shimoda, M.; Takada, Y. Integrin Binding to the Trimeric Interface of CD40L Plays a Critical Role in CD40/CD40L Signaling. J. Immunol. 2019, 203, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.; May, A.E.; Bültmann, A.; Gawaz, M. ADAM 15 is an adhesion receptor for platelet GPIIb-IIIa and induces platelet activation. Thromb. Haemost. 2005, 94, 555–561. [Google Scholar] [CrossRef]
- Han, J.; Lim, C.J.; Watanabe, N.; Soriani, A.; Ratnikov, B.; Calderwood, D.A.; Puzon-McLaughlin, W.; Lafuente, E.M.; Boussiotis, V.A.; Shattil, S.J.; et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr. Biol. 2006, 16, 1796–1806. [Google Scholar] [CrossRef]
- Presta, M.; Andrés, G.; Leali, D.; Dell’Era, P.; Ronca, R.; Dell’Era, P.; Ronca, R. Inflammatory cells and chemokines sustain FGF2-induced angiogenesis. Eur. Cytokine Netw. 2009, 20, 39–50. [Google Scholar] [CrossRef]
- Sun, L.-L.; Lei, F.-R.; Jiang, X.-D.; Du, X.-L.; Xiao, L.; Li, W.-D.; Li, X.-Q. LncRNA GUSBP5-AS promotes EPC migration and angiogenesis and deep vein thrombosis resolution by regulating FGF2 and MMP2/9 through the miR-223-3p/FOXO1/Akt pathway. Aging 2020, 12, 4506–4526. [Google Scholar] [CrossRef]
- Fan, L.; Ding, L.; Lan, J.; Niu, J.; He, Y.; Song, L. Fibroblast Growth Factor-1 Improves Insulin Resistance via Repression of JNK-Mediated Inflammation. Front. Pharmacol. 2019, 10, 1478. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.M.; Jonker, J.W.; Ahmadian, M.; Goetz, R.; Lackey, D.; Osborn, O.; Huang, Z.; Liu, W.; Yoshihara, E.; van Dijk, T.H.; et al. Evans Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2014, 513, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Qin, J. LINC00659 exacerbates endothelial progenitor cell dysfunction in deep vein thrombosis of the lower extremities by activating DNMT3A-mediated FGF1 promoter methylation. Thromb. J. 2023, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Zhang, X.-P.; Medved, L.; Takada, Y. Specific binding of integrin alpha v beta 3 to the fibrinogen gamma and alpha E chain C-terminal domains. Biochemistry 1999, 38, 5872–5877. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Malcolm, B.A. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 1999, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Ieguchi, K.; Fujita, M.; Ma, Z.; Davari, P.; Taniguchi, Y.; Sekiguchi, K.; Wang, B.; Takada, Y.K.; Takada, Y. Direct binding of the EGF-like domain of neuregulin-1 to integrins (αvβ3 and α6β4) is involved in neuregulin-1/ErbB signaling. J. Biol. Chem. 2010, 285, 31388–31398. [Google Scholar] [CrossRef]
- Saegusa, J.; Akakura, N.; Wu, C.-Y.; Hoogland, C.; Ma, Z.; Lam, K.S.; Liu, F.-T.; Takada, Y.K.; Takada, Y. Pro-inflammatory secretory phospholipase A2 type IIA binds to integrins alphavbeta3 and alpha4beta1 and induces proliferation of monocytic cells in an integrin-dependent manner. J. Biol. Chem. 2008, 283, 26107–26115. [Google Scholar] [CrossRef] [PubMed]
- Gailit, J.; Ruoslahti, E. Regulation of the fibronectin receptor affinity by divalent cations. J. Biol. Chem. 1988, 263, 12927–12932. [Google Scholar] [CrossRef]
- Mould, A.P.; Akiyama, S.K.; Humphries, M.J. Regulation of integrin alpha 5 beta 1-fibronectin interactions by divalent cations. Evidence for distinct classes of binding sites for Mn2+, Mg2+, and Ca2+. J. Biol. Chem. 1995, 270, 26270–26277. [Google Scholar] [CrossRef]
- Altieri, D.C. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J. Immunol. 1991, 147, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Elices, M.J.; Urry, L.A.; Hemler, M.E. Receptor functions for the integrin VLA-3: Fibronectin, collagen, and laminin binding are differentially influenced by Arg-Gly-Asp peptide and by divalent cations. J. Cell Biol. 1991, 112, 169–181. [Google Scholar] [CrossRef]
- André, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-derived CD40L: The switch-hitting player of cardiovascular disease. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef]
- Prasad, K.S.S.; Andre, P.; Yan, Y.; Phillips, D.R. The platelet CD40L/GP IIb-IIIa axis in atherothrombotic disease. Curr. Opin. Hematol. 2003, 10, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Merhi, Y. Implication of Platelets in Immuno-Thrombosis and Thrombo-Inflammation. Front. Cardiovasc. Med. 2022, 9, 863846. [Google Scholar] [CrossRef] [PubMed]
- Wienkamp, A.-K.; Erpenbeck, L.; Rossaint, J. Platelets in the NETworks interweaving inflammation and thrombosis. Front. Immunol. 2022, 13, 953129. [Google Scholar] [CrossRef]
- Sonmez, O.; Sonmez, M. Role of platelets in immune system and inflammation. Porto Biomed. J. 2017, 2, 311–314. [Google Scholar] [CrossRef]
- Liang, G.; Song, L.; Chen, Z.; Qian, Y.; Xie, J.; Zhao, L.; Lin, Q.; Zhu, G.; Tan, Y.; Li, X.; et al. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 2018, 93, 95–109. [Google Scholar] [CrossRef]
- Liu, W.; Struik, D.; Nies, V.J.M.; Jurdzinski, A.; Harkema, L.; de Bruin, A.; Verkade, H.J.; Downes, M.; Evans, R.M.; van Zutphen, T.; et al. Effective treatment of steatosis and steatohepatitis by fibroblast growth factor 1 in mouse models of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2016, 113, 2288–2293. [Google Scholar] [CrossRef]
- Gasser, E.; Moutos, C.P.; Downes, M.; Evans, R.M. FGF1—A new weapon to control type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2017, 13, 599–609. [Google Scholar] [CrossRef]
- Sancar, G.; Liu, S.; Gasser, E.; Alvarez, J.G.; Moutos, C.; Kim, K.; van Zutphen, T.; Wang, Y.; Huddy, T.F.; Ross, B.; et al. FGF1 and insulin control lipolysis by convergent pathways. Cell Metab. 2022, 34, 171–183 e176. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Engleka, K.; Zhan, X.; Tokita, Y.; Forough, R.; Roeder, D.; Jackson, A.; Maier, J.A.M.; Hla, T.; Maciag, T. Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science 1990, 249, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Hu, J.; Huang, W.; Ye, S.; Han, F.; Du, J.; Shao, M.; Guo, R.; Lin, J.; Zhao, Y.; et al. Non-Mitogenic Fibroblast Growth Factor 1 Enhanced Angiogenesis Following Ischemic Stroke by Regulating the Sphingosine-1-Phosphate 1 Pathway. Front. Pharmacol. 2020, 11, 59. [Google Scholar] [CrossRef]
- Szlachcic, A.; Sochacka, M.; Czyrek, A.; Opalinski, L.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Low Stability of Integrin-Binding Deficient Mutant of FGF1 Restricts Its Biological Activity. Cells 2019, 8, 899. [Google Scholar] [CrossRef] [PubMed]
- Billheimer, J.T.; He, B.; Spitz, S.M.; Stern, A.M.; Seiffert, D. Effects of glycoprotein IIb/IIIa antagonists on platelet activation: Development of a transfer method to mimic peak to trough receptor occupancy. Thromb. Res. 2002, 107, 303–317. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FGF2 (1BFG) | αv (1JV2) | β3 |
|---|---|---|
| Asp-19, Arg33, His35, Pro36, Asp37, Gly38, Arg39, Val43, Glu45, Lys46, Ser47, Asp48, Pro49, His50, Gln56, Lys66, Val68, Ser69, Ala70, Asn71, Arg72, Tyr73, Lys77, Arg81, Leu83, Ala84, Ser85, Lys86, Ser87, Val88, Thr89, Asp89, Asp90, Phe93, Lys110 | Glu15, Asn44, Gly49, Ile50, Val51, Glu52, Asn77, Asp83, Phe88, Ser90, His91, Arg122 | Pro160, Val161, Ser162, Met165, Ile167, Ser168, Glu171, Glu174, Asn175, Pro186, Met187, Lys235, Val275, Gly276, Ser277, Asp278, His280, Tyr281, Ser282, Ala283, Thr285, Thr286 |
| FGF1 (1AXM) | αv (1JV2) | β3 |
|---|---|---|
| Asn18, Gly19, Gly20, His21, Asp28, Gly29, Thr30, Val31, Asp32, Gly33, Arg35, Asp68, Thr69, Asp70, Leu72, Leu73, Glu81, Glu82, Lys101, Lys105, Trp107, Leu111, Lys112, Lys113, Asn114, Gly115, Ser116, Cys117, Lys118, Arg119, Pro121, Arg122, Thr123, His124, Tyr125, Gly126, Gln127, Lys128 | Asn44, Gly49, Ile50, Val51, Glu52, Asp79, Ala81, Lys82, Asp83, Asp84, Pro85, Phe88, Ser90, His91, His113, Gln120, Arg122 | Pro160, Val161, Ser162, Met165, Ile167, Ser168, Pro169, Pro170, Glu171, Ala172, Glu174, Asn175, Pro186, Met187, Phe188, Gly276, Ser277, Asp278, His280, Tyr281, Ser282, Thr285 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takada, Y.K.; Wu, X.; Wei, D.; Hwang, S.; Takada, Y. FGF1 Suppresses Allosteric Activation of β3 Integrins by FGF2: A Potential Mechanism of Anti-Inflammatory and Anti-Thrombotic Action of FGF1. Biomolecules 2024, 14, 888. https://doi.org/10.3390/biom14080888
Takada YK, Wu X, Wei D, Hwang S, Takada Y. FGF1 Suppresses Allosteric Activation of β3 Integrins by FGF2: A Potential Mechanism of Anti-Inflammatory and Anti-Thrombotic Action of FGF1. Biomolecules. 2024; 14(8):888. https://doi.org/10.3390/biom14080888
Chicago/Turabian StyleTakada, Yoko K., Xuesong Wu, David Wei, Samuel Hwang, and Yoshikazu Takada. 2024. "FGF1 Suppresses Allosteric Activation of β3 Integrins by FGF2: A Potential Mechanism of Anti-Inflammatory and Anti-Thrombotic Action of FGF1" Biomolecules 14, no. 8: 888. https://doi.org/10.3390/biom14080888