
A Novel Workflow for In Silico Prediction of Bioactive Peptides: An Exploration of Solanum lycopersicum By-Products

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Methods
2.1. Genome Mining and Protein Selection
2.2. In Silico Digestion
2.3. Bioactivity Peptide Prediction
2.4. Filtering
2.4.1. Screening for Toxicity, Bitterness, and Stability
2.4.2. Bioactivity Evaluation
2.5. Peptides Structure Prediction
2.6. Molecular Docking
2.7. Energy Calculation and Interaction Analysis
2.7.1. Molecular Dynamics for Re-Scoring and Energy Calculation
2.7.2. Interaction Analysis
3. Results
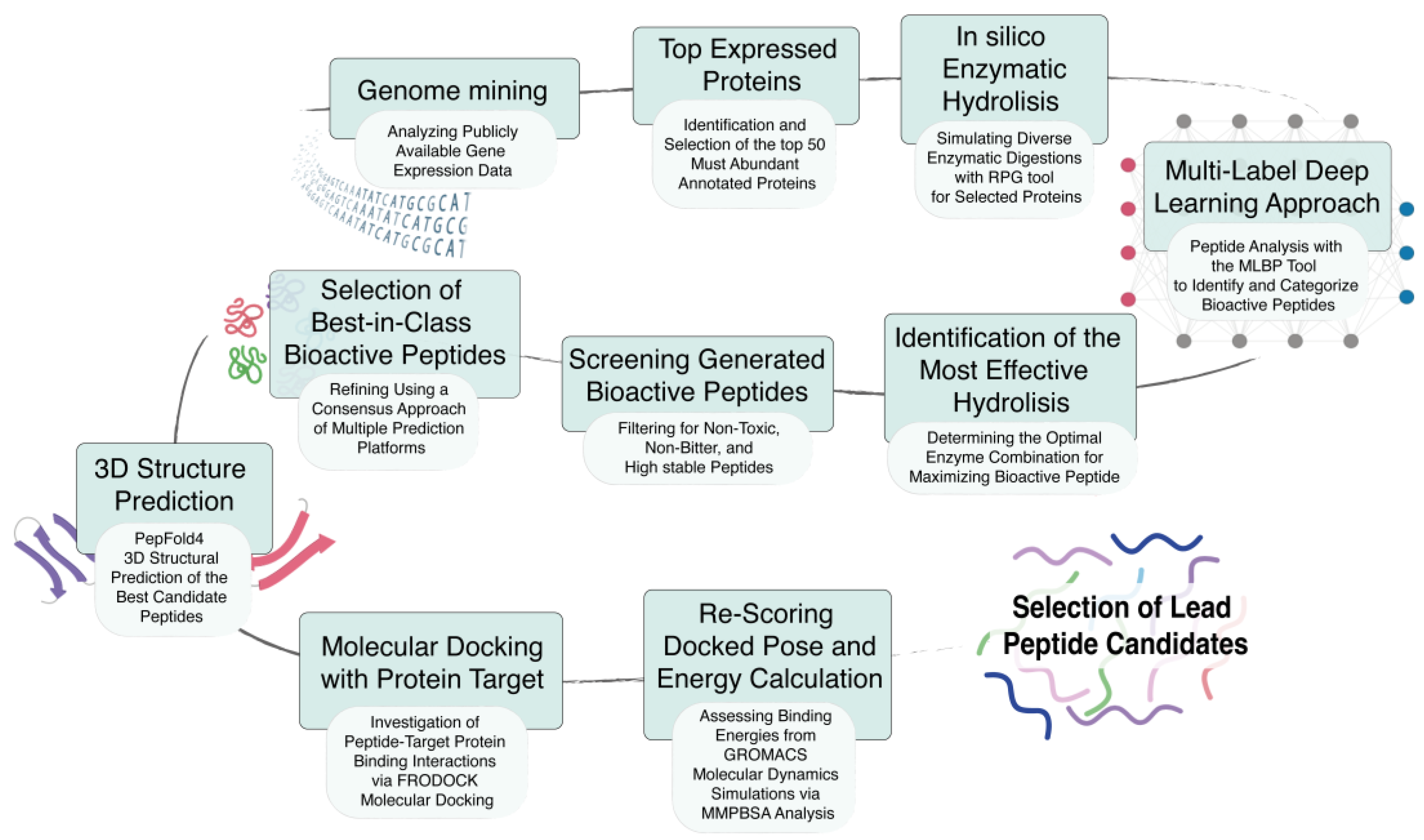
3.1. Workflow for the Identification and Evaluation of Bioactive Peptides
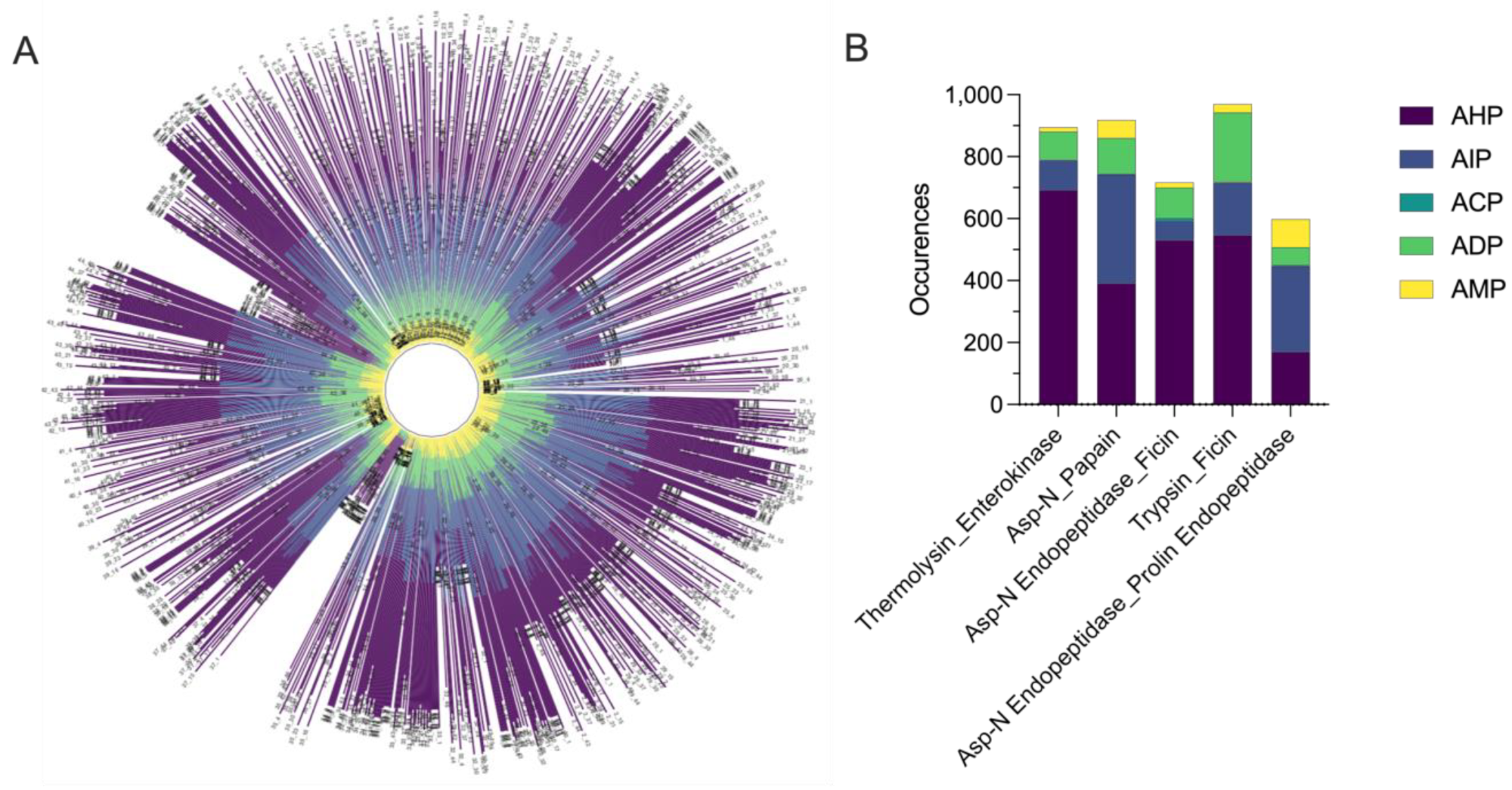
3.2. Simulated Enzymatic Hydrolysis
3.3. Prediction of Solanum lycopersicum Bio-Active Peptides (BAPs)
3.4. Prospect Solanum lycopersicum Bioactive Peptides
3.4.1. Anticancer Peptides (ACPs)
3.4.2. Antidiabetic Peptides (ADPs)
3.4.3. Antihypertensive Peptides (AHPs)
3.4.4. Anti-Inflammatory Peptides (AIPs)
3.4.5. Antimicrobial Peptides (AMPs)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Czelej, M.; Garbacz, K.; Czernecki, T.; Wawrzykowski, J.; Waśko, A. Protein Hydrolysates Derived from Animals and Plants—A Review of Production Methods and Antioxidant Activity. Foods 2022, 11, 1953. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, N.; Chen, F.; Zhang, J.; Sun, X.; Xu, L.; Fang, F. Review on the Release Mechanism and Debittering Technology of Bitter Peptides from Protein Hydrolysates. Compr. Rev. Food Sci. Food Saf. 2022, 21, 5153–5170. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Wu, N.; Tang, S.; Xiao, N.; Jiang, Y.; Tu, Y.; Xu, M. Industrial Application of Protein Hydrolysates in Food. J. Agric. Food Chem. 2023, 71, 1788–1801. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.-P.; Liu, H.-Q.; Ye, Z.-W.; Zheng, Q.-W.; Zou, Y.; Wei, T.; Guo, L.-Q.; Lin, J.-F. The Beneficial Potential of Protein Hydrolysates as Prebiotic for Probiotics and Its Biological Activity: A Review. Crit. Rev. Food Sci. Nutr. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Klaassen Kamdar, A.; Labuza, T.P. Storage Stability of Food Protein Hydrolysates—A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1169–1192. [Google Scholar] [CrossRef] [PubMed]
- Sivaperumal, P.; Kamala, K.; Rajaram, R. Chapter Eight—Bioremediation of Industrial Waste Through Enzyme Producing Marine Microorganisms. In Advances in Food and Nutrition Research, Marine Enzymes Biotechnology: Production and Industrial Applications, Part III-Application of Marine Enzymes; Kim, S.-K., Toldrá, F., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 80, pp. 165–179. [Google Scholar]
- Asaithambi, N.; Singha, P.; Singh, S.K. Recent Application of Protein Hydrolysates in Food Texture Modification. Crit. Rev. Food Sci. Nutr. 2022, 63, 10412–10443. [Google Scholar] [CrossRef]
- Nikoo, M.; Regenstein, J.M.; Yasemi, M. Protein Hydrolysates from Fishery Processing By-Products: Production, Characteristics, Food Applications, and Challenges. Foods 2023, 12, 4470. [Google Scholar] [CrossRef]
- Cesaretti, A.; Montegiove, N.; Calzoni, E.; Leonardi, L.; Emiliani, C. Protein hydrolysates: From agricultural waste biomasses to high added-value products (minireview). AgroLife Sci. J. 2020, 9, 79–87. [Google Scholar]
- Marquez-Rios, E.; Del-Toro-Sanchez, C.L. Antioxidant Peptides from Terrestrial and Aquatic Plants Against Cancer. Curr. Protein Pept. Sci. 2018, 19, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yuan, N.; Guo, W.; Chai, Y.; Song, Y.; Zhao, Y.; Zeng, M.; Wu, H. Antioxidant and Anti-Inflammatory Protective Effects of Yellowtail (Seriola quinqueradiata) Milt Hydrolysates on Human Intestinal Epithelial Cells In Vitro and Dextran Sodium Sulphate-Induced Mouse Colitis In Vivo. Food Funct. 2022, 13, 9169–9182. [Google Scholar] [CrossRef]
- Akbarmehr, A.; Peighambardoust, S.H.; Ghanbarzadeh, B.; Sarabandi, K. Physicochemical, Antioxidant, Antimicrobial, and in Vitro Cytotoxic Activities of Corn Pollen Protein Hydrolysates Obtained by Different Peptidases. Food Sci. Nutr. 2023, 11, 2403–2417. [Google Scholar] [CrossRef]
- Bin Hafeez, A.; Jiang, X.; Bergen, P.J.; Zhu, Y. Antimicrobial Peptides: An Update on Classifications and Databases. Int. J. Mol. Sci. 2021, 22, 11691. [Google Scholar] [CrossRef] [PubMed]
- Chalamaiah, M.; Yu, W.; Wu, J. Immunomodulatory and Anticancer Protein Hydrolysates (Peptides) from Food Proteins: A Review. Food Chem. 2018, 245, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Kiewiet, M.B.G.; Faas, M.M.; De Vos, P. Immunomodulatory Protein Hydrolysates and Their Application. Nutrients 2018, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- UG, Y.; Bhat, I.; Karunasagar, I.; BS, M. Antihypertensive Activity of Fish Protein Hydrolysates and Its Peptides. Crit. Rev. Food Sci. Nutr. 2019, 59, 2363–2374. [Google Scholar]
- Correia-da-Silva, M.; Sousa, E.; Pinto, M.M.M.; Kijjoa, A. Anticancer and Cancer Preventive Compounds from Edible Marine Organisms. Semin. Cancer Biol. 2017, 46, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Jimenez, G.-M.; Burgos-Hernandez, A.; Ezquerra-Brauer, J.-M. Bioactive Peptides and Depsipeptides with Anticancer Potential: Sources from Marine Animals. Mar. Drugs 2012, 10, 963–986. [Google Scholar] [CrossRef] [PubMed]
- Nasri, R.; Abdelhedi, O.; Jemil, I.; Amor, I.B.; Elfeki, A.; Gargouri, J.; Boualga, A.; Karra-Châabouni, M.; Nasri, M. Preventive Effect of Goby Fish Protein Hydrolysates on Hyperlipidemia and Cardiovascular Disease in Wistar Rats Fed a High-Fat/Fructose Diet. RSC Adv. 2018, 8, 9383–9393. [Google Scholar] [CrossRef] [PubMed]
- Ktari, N.; Mnafgui, K.; Nasri, R.; Hamden, K.; Bkhairia, I.; Hadj, A.B.; Boudaouara, T.; Elfeki, A.; Nasri, M. Hypoglycemic and Hypolipidemic Effects of Protein Hydrolysates from Zebra Blenny (Salaria basilisca) in Alloxan-Induced Diabetic Rats. Food Funct. 2013, 4, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Megías, C.; Pedroche, J.; del Mar Yust, M.; Alaiz, M.; Girón-Calle, J.; Millán, F.; Vioque, J. Sunflower Protein Hydrolysates Reduce Cholesterol Micellar Solubility. Plant Foods Hum. Nutr. 2009, 64, 86–93. [Google Scholar] [CrossRef]
- Abeer, M.M.; Trajkovic, S.; Brayden, D.J. Measuring the Oral Bioavailability of Protein Hydrolysates Derived from Food Sources: A Critical Review of Current Bioassays. Biomed. Pharmacother. 2021, 144, 112275. [Google Scholar] [CrossRef]
- Gallego, M.; Grootaert, C.; Mora, L.; Aristoy, M.C.; Van Camp, J.; Toldrá, F. Transepithelial Transport of Dry-Cured Ham Peptides with ACE Inhibitory Activity through a Caco-2 Cell Monolayer. J. Funct. Foods 2016, 21, 388–395. [Google Scholar] [CrossRef]
- Xu, Q.; Hong, H.; Wu, J.; Yan, X. Bioavailability of Bioactive Peptides Derived from Food Proteins across the Intestinal Epithelial Membrane: A Review. Trends Food Sci. Technol. 2019, 86, 399–411. [Google Scholar] [CrossRef]
- Vats, S.; Bansal, R.; Rana, N.; Kumawat, S.; Bhatt, V.; Jadhav, P.; Kale, V.; Sathe, A.; Sonah, H.; Jugdaohsingh, R.; et al. Unexplored Nutritive Potential of Tomato to Combat Global Malnutrition. Crit. Rev. Food. Sci. Nutr. 2022, 62, 1003–1034. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pan, I.; Ibañez, F.C.; Virseda, P.; Arozarena, I.; Beriain, M.J. Stabilization and Valorization of Tomato Byproduct: A Case Study for the Bakery Industry. J. Food Sci. 2023, 88, 4483–4494. [Google Scholar] [CrossRef] [PubMed]
- Kaboré, K.; Konaté, K.; Sanou, A.; Dakuyo, R.; Sama, H.; Santara, B.; Compaoré, E.W.R.; Dicko, M.H. Tomato By-Products, a Source of Nutrients for the Prevention and Reduction of Malnutrition. Nutrients 2022, 14, 2871. [Google Scholar] [CrossRef] [PubMed]
- López-Yerena, A.; Domínguez-López, I.; Abuhabib, M.M.; Lamuela-Raventós, R.M.; Vallverdú-Queralt, A.; Pérez, M. Tomato Wastes and By-Products: Upcoming Sources of Polyphenols and Carotenoids for Food, Nutraceutical, and Pharma Applications. Crit. Rev. Food Sci. Nutr. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Paulino, S.-L.J.; Adrián, Á.-T.G.; Gabriela, E.-A.L.; Maribel, V.-M.; Sergio, M.-G. Nutraceutical Potential of Flours from Tomato By-Product and Tomato Field Waste. J. Food Sci. Technol. 2020, 57, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Santagata, R.; Ripa, M.; Genovese, A.; Ulgiati, S. Food Waste Recovery Pathways: Challenges and Opportunities for an Emerging Bio-Based Circular Economy. A Systematic Review and an Assessment. J. Clean. Prod. 2021, 286, 125490. [Google Scholar] [CrossRef]
- Nasri, M. Protein Hydrolysates and Biopeptides: Production, Biological Activities, and Applications in Foods and Health Benefits. A Review. In Advances in Food and Nutrition Research; Academic Press: Cambridge, MA, USA, 2017; Volume 81, pp. 109–159. [Google Scholar]
- Heffernan, S.; Giblin, L.; O’Brien, N. Assessment of the Biological Activity of Fish Muscle Protein Hydrolysates Using In Vitro Model Systems. Food Chem. 2021, 359, 129852. [Google Scholar] [CrossRef]
- Yao, H.; Yang, J.; Zhan, J.; Lu, Q.; Su, M.; Jiang, Y. Preparation, Amino Acid Composition, and in Vitro Antioxidant Activity of Okra Seed Meal Protein Hydrolysates. Food Sci. Nutr. 2021, 9, 3059–3070. [Google Scholar] [CrossRef]
- Mirzaee, H.; Ahmadi Gavlighi, H.; Nikoo, M.; Udenigwe, C.C.; Khodaiyan, F. Relation of Amino Acid Composition, Hydrophobicity, and Molecular Weight with Antidiabetic, Antihypertensive, and Antioxidant Properties of Mixtures of Corn Gluten and Soy Protein Hydrolysates. Food Sci. Nutr. 2022, 11, 1257–1271. [Google Scholar] [CrossRef]
- Calzoni, E.; Cesaretti, A.; Tacchi, S.; Caponi, S.; Pellegrino, R.M.; Luzi, F.; Cottone, F.; Fioretto, D.; Emiliani, C.; Di Michele, A. Covalent Immobilization of Proteases on Polylactic Acid for Proteins Hydrolysis and Waste Biomass Protein Content Valorization. Catalysts 2021, 11, 167. [Google Scholar] [CrossRef]
- Manzoor, M.; Singh, J.; Bhat, Z.F.; Jaglan, S. Multifunctional Apple Seed Protein Hydrolysates: Impact of Enzymolysis on the Biochemical, Techno-Functional and in Vitro α-Glucosidase, Pancreatic Lipase and Angiotensin-Converting Enzyme Inhibition Activities. Int. J. Biol. Macromol. 2024, 257, 128553. [Google Scholar] [CrossRef] [PubMed]
- Mohd Salim, M.A.S.; Gan, C.-Y. Dual-Function Peptides Derived from Egg White Ovalbumin: Bioinformatics Identification with Validation Using in Vitro Assay. J. Funct. Foods 2020, 64, 103618. [Google Scholar] [CrossRef]
- Desmarchelier, C.; Borel, P. Overview of Carotenoid Bioavailability Determinants: From Dietary Factors to Host Genetic Variations. Trends Food Sci. Technol. 2017, 69, 270–280. [Google Scholar] [CrossRef]
- Manzoor, M.; Singh, J.; Gani, A. Exploration of Bioactive Peptides from Various Origin as Promising Nutraceutical Treasures: In Vitro, in Silico and in Vivo Studies. Food Chem. 2022, 373, 131395. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.K.; Nataraj, B.H. Revisiting Therapeutic and Toxicological Fingerprints of Milk-Derived Bioactive Peptides: An Overview. Food Biosci. 2020, 38, 100771. [Google Scholar] [CrossRef]
- Wang, T.-Y.; Hsieh, C.-H.; Hung, C.-C.; Jao, C.-L.; Lin, P.-Y.; Hsieh, Y.-L.; Hsu, K.-C. A Study to Evaluate the Potential of an in Silico Approach for Predicting Dipeptidyl Peptidase-IV Inhibitory Activity in Vitro of Protein Hydrolysates. Food Chem. 2017, 234, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Coelho, L.P.; Santos-Júnior, C.D.; de la Fuente-Nunez, C. Challenges in Computational Discovery of Bioactive Peptides in ’omics Data. Proteomics 2024, 24, 2300105. [Google Scholar] [CrossRef]
- Bizzotto, E.; Zampieri, G.; Treu, L.; Filannino, P.; Cagno, R.D.; Campanaro, S. Classification of Bioactive Peptides: A Systematic Benchmark of Models and Encodings. Comput. Struct. Biotechnol. J. 2024, 23, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Comer, J.; Li, Y. Bioinformatics Approaches to Discovering Food-Derived Bioactive Peptides: Reviews and Perspectives. TrAC Trends Anal. Chem. 2023, 162, 117051. [Google Scholar] [CrossRef]
- Ozaki, S.; Ogata, Y.; Suda, K.; Kurabayashi, A.; Suzuki, T.; Yamamoto, N.; Iijima, Y.; Tsugane, T.; Fujii, T.; Konishi, C.; et al. Coexpression Analysis of Tomato Genes and Experimental Verification of Coordinated Expression of Genes Found in a Functionally Enriched Coexpression Module. DNA Res. 2010, 17, 105–116. [Google Scholar] [CrossRef]
- Maillet, N. Rapid Peptides Generator: Fast and Efficient in Silico Protein Digestion. NAR Genom. Bioinform. 2020, 2, lqz004. [Google Scholar] [CrossRef]
- Tang, W.; Dai, R.; Yan, W.; Zhang, W.; Bin, Y.; Xia, E.; Xia, J. Identifying Multi-Functional Bioactive Peptide Functions Using Multi-Label Deep Learning. Brief. Bioinform. 2022, 23, bbab414. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.S.; Arora, A.; Choudhury, S.; Tijare, P.; Raghava, G.P.S. ToxinPred 3.0: An Improved Method for Predicting the Toxicity of Peptides. Comput. Biol. Med. 2024, 179, 108926. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Yana, J.; Schaduangrat, N.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iBitter-SCM: Identification and Characterization of Bitter Peptides Using a Scoring Card Method with Propensity Scores of Dipeptides. Genomics 2020, 112, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Singh, S.; Mehta, A.; Agrawal, P.; Raghava, G.P.S. In Silico Approaches for Predicting the Half-Life of Natural and Modified Peptides in Blood. PLoS ONE 2018, 13, e0196829. [Google Scholar] [CrossRef] [PubMed]
- Gülseren, İ.; Vahapoglu, B. The Stability of Food Bioactive Peptides in Blood: An Overview. Int. J. Pept. Res. Ther. 2022, 28, 2. [Google Scholar] [CrossRef]
- Sharma, A.; Singla, D.; Rashid, M.; Raghava, G.P.S. Designing of Peptides with Desired Half-Life in Intestine-like Environment. BMC Bioinform. 2014, 15, 282. [Google Scholar] [CrossRef] [PubMed]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.T.; Park, H.W.; Pitti, T.; Madhavan, T.; Jeon, Y.-J.; Manavalan, B. MLACP 2.0: An Updated Machine Learning Tool for Anticancer Peptide Prediction. Comput. Struct. Biotechnol. J. 2022, 20, 4473–4480. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, T.; Li, X.; Zhu, Y.; Chen, S.; Huang, J.; Zhou, F.; Liu, H. ACPPfel: Explainable Deep Ensemble Learning for Anticancer Peptides Prediction Based on Feature Optimization. Front. Genet. 2024, 15, 1352504. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Singh Chauhan, J.; Nagpal, G.; Kumar, R.; Sharma, M.; Raghava, G.P.S. An in Silico Platform for Predicting, Screening and Designing of Antihypertensive Peptides. Sci. Rep. 2015, 5, 12512. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Kanthawong, S.; Nantasenamat, C.; Hasan, M.d.M.; Shoombuatong, W. iDPPIV-SCM: A Sequence-Based Predictor for Identifying and Analyzing Dipeptidyl Peptidase IV (DPP-IV) Inhibitory Peptides Using a Scoring Card Method. J. Proteome Res. 2020, 19, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-Inflammatory Peptides Using Random Forest. Front. Pharmacol. 2018, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A Database on Sequences, Structures and Signatures of Antimicrobial Peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Rey, J.; Murail, S.; de Vries, S.; Derreumaux, P.; Tuffery, P. PEP-FOLD4: A pH-Dependent Force Field for Peptide Structure Prediction in Aqueous Solution. Nucleic Acids Res. 2023, 51, W432–W437. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.J.; Yin, Y.W.; Ma, Y.Q.; Ding, H.M. Improving the Performance of MM/PBSA in Protein–Protein Interactions via the Screening Electrostatic Energy. J. Chem. Inf. Model. 2021, 61, 2454–2462. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Sheng, Y.J.; Ma, Y.Q.; Ding, H.M. Assessing the Performance of Screening MM/PBSA in Protein–Ligand Interactions. J. Phys. Chem. B 2022, 126, 1700–1708. [Google Scholar] [CrossRef]
- Morena, F.; Argentati, C.; Tortorella, I.; Emiliani, C.; Martino, S. De Novo ssRNA Aptamers against the SARS-CoV-2 Main Protease: In Silico Design and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2021, 22, 6874. [Google Scholar] [CrossRef]
- Martarelli, N.; Capurro, M.; Mansour, G.; Jahromi, R.V.; Stella, A.; Rossi, R.; Longetti, E.; Bigerna, B.; Gentili, M.; Rosseto, A.; et al. Artificial Intelligence-Powered Molecular Docking and Steered Molecular Dynamics for Accurate scFv Selection of Anti-CD30 Chimeric Antigen Receptors. Int. J. Mol. Sci. 2024, 25, 7231. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A Web Server to Predict and Analyze the Protein–Protein Complex Based on Computational Docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; FitzGerald, R.J. Features of Dipeptidyl Peptidase IV (DPP-IV) Inhibitory Peptides from Dietary Proteins. J. Food Biochem. 2019, 43, e12451. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Chen, L.; Winter, M.B.; Lin, Y.-L.; Yang, Y.; Shapovalova, M.; Smith, P.M.; Liu, C.; Li, F.; LeBeau, A.M. The Rational Design of Therapeutic Peptides for Aminopeptidase N Using a Substrate-Based Approach. Sci. Rep. 2017, 7, 1424. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Guha, S.; Majumder, K. Food-Derived Bioactive Peptides in Human Health: Challenges and Opportunities. Nutrients 2018, 10, 1738. [Google Scholar] [CrossRef] [PubMed]
- Solanki, D.; Hati, S.; Sakure, A. In Silico and In Vitro Analysis of Novel Angiotensin I-Converting Enzyme (ACE) Inhibitory Bioactive Peptides Derived from Fermented Camel Milk (Camelus dromedarius). Int. J. Pept. Res. Ther. 2017, 23, 441–459. [Google Scholar] [CrossRef]
- Toldrá, F.; Reig, M.; Aristoy, M.-C.; Mora, L. Generation of Bioactive Peptides during Food Processing. Food Chem. 2018, 267, 395–404. [Google Scholar] [CrossRef]
- Martini, S.; Conte, A.; Tagliazucchi, D. Effect of Ripening and in Vitro Digestion on the Evolution and Fate of Bioactive Peptides in Parmigiano-Reggiano Cheese. Int. Dairy J. 2020, 105, 104668. [Google Scholar] [CrossRef]
- Zielińska, E.; Baraniak, B.; Karaś, M. Identification of Antioxidant and Anti-Inflammatory Peptides Obtained by Simulated Gastrointestinal Digestion of Three Edible Insects Species (Gryllodes sigillatus, Tenebrio molitor, Schistocerca gragaria). Int. J. Food Sci. Technol. 2018, 53, 2542–2551. [Google Scholar] [CrossRef]
- Szerszunowicz, I.; Kozicki, S. Plant-Derived Proteins and Peptides as Potential Immunomodulators. Molecules 2023, 29, 209. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; You, M.; Song, H.; Gong, L.; Pan, W. Investigation of umami and kokumi taste-active components in bovine bone marrow extract produced during enzymatic hydrolysis and Maillard reaction. Int. J. Food Sci. Technol. 2018, 53, 2465–2481. [Google Scholar] [CrossRef]
- Daroit, D.J.; Brandelli, A. In Vivo Bioactivities of Food Protein-Derived Peptides—A Current Review. Curr. Opin. Food Sci. 2021, 39, 120–129. [Google Scholar] [CrossRef]
- Munir, M.; Nadeem, M.; Mahmood Qureshi, T.; Gamlath, C.J.; Martin, G.J.O.; Hemar, Y.; Ashokkumar, M. Effect of Sonication, Microwaves and High-Pressure Processing on ACE-Inhibitory Activity and Antioxidant Potential of Cheddar Cheese during Ripening. Ultrason. Sonochem. 2020, 67, 105140. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhong, X.; Luan, C.; Wen, N.; Shi, C.; Liu, S.; Xu, Y.; He, Q.; Wu, Y.; Yang, J. Simultaneous Ultrasound and Microwave Application in Myosin-Chlorogenic Acid Conjugation: Unlocking Enhanced Emulsion Stability. Food Chem. 2024, 21, 101149. [Google Scholar] [CrossRef] [PubMed]
- Chandimali, N.; Bak, S.-G.; Hyun Park, E.; Lim, H.-J.; Won, Y.-S.; Kim, B.; Lee, S.-J. Bioactive Peptides Derived from Duck Products and By-Products as Functional Food Ingredients. J. Funct. Foods 2024, 113, 105953. [Google Scholar] [CrossRef]
- Peredo-Lovillo, A.; Hernández-Mendoza, A.; Vallejo-Cordoba, B.; Romero-Luna, H.E. Conventional and in Silico Approaches to Select Promising Food-Derived Bioactive Peptides: A Review. Food Chem. 2021, 13, 100183. [Google Scholar] [CrossRef]
- Polak-Berecka, M.; Michalak-Tomczyk, M.; Skrzypczak, K.; Michalak, K.; Rachwał, K.; Waśko, A. Potential Biological Activities of Peptides Generated during Casein Proteolysis by Curly Kale (Brassica oleracea L. var. sabellica L.) Leaf Extract: An In Silico Preliminary Study. Foods 2021, 10, 2877. [Google Scholar] [CrossRef]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M. Bioinformatics and Bioactive Peptides from Foods: Do They Work Together? Adv. Food Nutr. Res. 2024, 108, 35–111. [Google Scholar] [CrossRef]
- Zhou, L.; Mendez, R.L.; Kwon, J.Y. In Silico Prospecting for Novel Bioactive Peptides from Seafoods: A Case Study on Pacific Oyster (Crassostrea gigas). Molecules 2023, 28, 651. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Tomar, M.; Bhuyan, D.J.; Punia, S.; Grasso, S.; Sá, A.G.A.; Carciofi, B.A.M.; Arrutia, F.; Changan, S.; Singh, S.; et al. Tomato (Solanum lycopersicum L.) Seed: A Review on Bioactives and Biomedical Activities. Biomed. Pharmacother. 2021, 142, 112018. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.C.; Rodrigues, A.S.; Teixeira, J.A.; Pintado, M.E. Integral Valorisation of Tomato By-Products towards Bioactive Compounds Recovery: Human Health Benefits. Food Chem. 2023, 410, 135319. [Google Scholar] [CrossRef] [PubMed]
- Zaky, A.A.; Simal-Gandara, J.; Eun, J.-B.; Shim, J.-H.; Abd El-Aty, A.M. Bioactivities, Applications, Safety, and Health Benefits of Bioactive Peptides from Food and By-Products: A Review. Front. Nutr. 2022, 8, 815640. [Google Scholar] [CrossRef]
- Jaglan, P.; Buttar, H.S.; Al-bawareed, O.A.; Chibisov, S. Chapter 24—Potential Health Benefits of Selected Fruits: Apples, Blueberries, Grapes, Guavas, Mangos, Pomegranates, and Tomatoes. In Functional Foods and Nutraceuticals in Metabolic and Non-Communicable Diseases; Singh, R.B., Watanabe, S., Isaza, A.A., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 359–370. ISBN 978-0-12-819815-5. [Google Scholar]
- Laranjeira, T.; Costa, A.; Faria-Silva, C.; Ribeiro, D.; De Oliveira, J.M.P.F.; Simões, S.; Ascenso, A. Sustainable Valorization of Tomato By-Products to Obtain Bioactive Compounds: Their Potential in Inflammation and Cancer Management. Molecules 2022, 27, 1701. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Bioactivity | Tool | Site | Ref. |
|---|---|---|---|
| ACP | ACPred | http://codes.bio/acpred/ (accessed on 8 March 2024) | [53] |
| MLACP2 | https://balalab-skku.org/mlacp2/ (accessed on 8 March 2024) | [54] | |
| ACPPfel | http://lmylab.online:5001/ (accessed on 8 March 2024) | [55] | |
| AHP | AHTpin | http://crdd.osdd.net/raghava/ahtpin (accessed on 8 March 2024) | [56] |
| ADP | iDPPIV-SCM | http://camt.pythonanywhere.com/iDPPIV-SCM (accessed on 8 March 2024) | [57] |
| AIP | AIPpred | http://211.239.150.230/AIPpred/AIPpredMethod.html/ (accessed on 8 March 2024) | [58] |
| AMP | CAMPR3 | http://www.camp3.bicnirrh.res.in/ (accessed on 8 March 2024) | [59] |
| Bioactive Class | Total | Toxic | Non-Toxic Non-Bitter High Stable |
|---|---|---|---|
| ACP | 10 | 6 | 4 |
| ADP | 187 | 36 | 49 |
| AHP | 692 | 144 | 220 |
| AIP | 351 | 9 | 66 |
| AMP | 91 | 15 | 13 |
| Peptide Sequence | Accession Number | Protein Name | Residues Number | MMGPBSA (kcal/mol ± SD) | Target Binding Residues | Peptide Binding Residues |
|---|---|---|---|---|---|---|
| MWKLPMFGCT | P05349 | Ribulose bisphosphate carboxylase small subunit, chloroplastic 4 | 10 | −52.98 ± 2.58 | G352 *, A353 *, R381, R442 | K3, P5, C9 |
| WKLPM | P05349 | Ribulose bisphosphate carboxylase small subunit, chloroplastic 4 | 5 | −45.91 ± 1.77 | Q213, E355 *, E389 *, Y477 * | W1, K2, L3 |
| Peptide Sequence | Accession Number | Protein Name | Residues Number | MMGPBSA (kcal/mol ± SD) | Target Binding Residues | Peptide Binding Residues |
|---|---|---|---|---|---|---|
| DLLNIFE | Q84K11 | Sine/threonine-Ptein phosphatase-5 | 7 | −41.36 ± 1.47 | R122, E202, E203, S206, Y544, R666 | D1, L2, N4, I5 |
| LMAALNLVG | P37222 | NADP-dependent malic enzyme, chloroplastic | 9 | −38.08± 1.52 | E202, E203, G629 | L1, N6 |
| HWLNTHAVIE | P38416 | Linoleate 9S-lipoxygenase B | 10 | −36.91 ± 3.16 | E202, E203, Y544, R666, N707 | H1, N4, T5, E10 |
| TFAFQAE | P36181 | Heat shock cognate Ptein 80 | 7 | −36.46 ± 5.99 | E202, E203, Y544, S624, S627, Y659, D660 | T1, Q5, E7 |
| QFVTFMK | Q6DUX3 | Translationally controlled tumor Protein homolog | 7 | −35.22 ± 6.75 | Y659, N706, N707, D736 | M6, K7 |
| QEFAHDFQAY | P05117 | Polygalacturonase-2 | 10 | −33.71 ± 4.20 | R557, K551, S627 * | F7, Q8, A9 |
| LPHPDGDQFG | P38416 | Linoleate 9S-lipoxygenase B | 10 | −33.24 ± 1.29 | R122, Y544, P547, K551, Y626, Y659 | L1, P2, H3, D5 |
| GWAPQVLLLS | K4D422 | UDP-Glycosyltransferase 73C4 | 10 | −31.93 ± 2.32 | R122, E202, S206 | G1, Q5 |
| LAQNNVMFE | NP_001294934 | Fructose-bisphosphate aldolase | 9 | −30.91 ± 1.43 | R122, E202, E203, C548, Y582 | L1, Q3, N5, E9 |
| EMIWDLLVS | Q40140 | Dartic Ptease | 9 | −30.7 ± 4.15 | K119, R557, N559, D736, E738 | E1, I3, L7, V8 |
| Peptide Sequence | Accession Number | Protein Name | Residues Number | MMGPBSA (kcal/mol ± SD) | Target Binding Residues | Peptide Binding Residues |
|---|---|---|---|---|---|---|
| MEMGESP | Q40158 | Metallothionein-like Ptein type 2 B | 7 | −60.77 ± 6.10 | Q886, E989, H992, E1016 *, K1112, H1114, Y1121 | M1, E2, E5, S6 |
| VEMQDVKYP | NP_001234021 | Polygalacturonase-2 precursor | 9 | −55.91 ± 3.41 | H992, Y999, R1007, Q1008, E1016 *, R1123 | V1, E2, M3, Q4, K7 |
| MEEVDVAPPQK | P28032 | Alcohol dehydrogenase 2 | 11 | −54.94 ± 6.25 | Y667, N671, N690, R729, Y740, E989 *, Y999, E1008, E1016 *, R1123, | E2, E3, D4, P9, Q10, K11 |
| ANQPLPDDDDEA | P38546 | GTP-binding nuclear Ptein Ran1 | 12 | −50.70 ± 10.09 | N671, R719, K723, D726, R729, Y818, Y965, R1123 | A1, N2, Q3, P4, D7, D8, D9 |
| VIPKENN | P30264 | Catalase isozyme 1 | 7 | −50.18 ± 7.84 | E767, Q886, D982, V984, V985, K1055, K1112, Y1124 | V1, K4, E5, N6, N7 |
| IDWKETPEPH | P30221 | Class I heat shock Protein | 10 | −44.58 ± 6.55 | D726, R729, Y829 *, D963, Y965 E1008, S1118 *, R1123 | I1, D2, W3, K4, E5, T6, E8, H10 |
| FEKGTHIPP | P37222 | NADP-dependent malic enzyme | 9 | −41.23 ± 1.85 | N671, A959, D963, E989 *, E1008, R1123 | F1, E2, K3, T5, P8 |
| FYQYNPDS | P29000 | Acid beta-fructofuranosidase | 8 | −36.80 ± 0.52 | Q886, A989, A959, S960, E989 *, E1016 *, Y1124, K1112, S1127 * | F1, Y2, Q3, Y4, N5, D7, S8 |
| VKVPEPT | P31542 | ATP-dependent Clp Ptease ATP-binding subunit ClpA homolog CD4B | 7 | −31.71 ± 1.87 | T667, N671, R729, E989, E1016 * | V1, K2, E5 |
| Peptide Sequence | Accession Number | Protein Name | Residues Number | MMGPBSA (kcal/mol ± SD) | Target Binding Residues | Peptide Binding Residues |
|---|---|---|---|---|---|---|
| PTKGSSVAIFGLGAVG-LAAAEGAR | P28032 | Alcohol dehydrogenase 2 | 24 | −50.68 ± 2.3 | F476 *, M480 | S6, I9 |
| DSSMAGYMSSKKTMEI-NPENSIM | P36181 | Heat shock cognate Ptein 80 | 23 | −47.66 ± 1.77 | S118, R381, K411, D412, P466, S469 | Y7, T13, M14, E15, N20, S21 |
| NQKNLHKRYAYQIVL-QTREMLR | Q84K13 | Sine/threonine-protein phosphatase 5 | 22 | −42.12 ± 3.52 | D460, F476 *, M480, N482 | R8, Y9, Q12, Q16 |
| DKRIFFTNKSYLPSQTP-SGVIR | AAB65766 | Lipoxygenase | 22 | −41.85 ± 1.58 | D98, L100, C115, F476, M480, N482 | K2, R3, F5, T7 |
| DTQPPRLPTKAVRVTA-EEVR | P54767 | Estimate decarboxylase | 20 | −32.76 ± 1.31 | R86, D412, E479 | K10, R20 |
| DETPELMPLSHVLATKLGAR | P43280 | S-adenosylmethionine synthase 1 | 20 | −32.72 ± 5.92 | D412, F476 *, | L9, S10, R20 |
| SAILATPSGERTMTSEQMVY | P08196 | Phytoene synthase 1, chloroplastic | 20 | −32.46 ± 3.72 | C115, P466, G470, E479 | T6, R11, Y20 |
| CGYSMNSIEGAAVSTIHITPE | ABQ42184 | S-adenosylmethionine decarboxylase Penzyme | 21 | −30.5 ± 0.99 | S118, R381 *, Q387 | N6, S7, E9 |
| Peptide Sequence | Accession Number | Protein Name | Residues Number | MMGPBSA (kcal/mol ± SD) | Target Binding Residues | Peptide Binding Residues |
|---|---|---|---|---|---|---|
| DGPNASYITPAAL | P08196 | Phytoene synthase 1, chloroplastic | 13 | −39.64 ± 0.64 | T111, G113, K114, T115 *, N151 *, H181, R187 *, D207, R334, H352, R382 | D1, N4, A5, S6, Y7, I8, A11 |
| DFGWGNPIFGGILKAISFTSFGVSVKN | Q6QLX4 | Alcohol acyl transferase | 27 | −34.96 ± 5.57 | Y45, E213, H217 | P7, K14 |
| DAGASKTYPQQAGTIRKGGHIVIKNRP | Q9AXQ6 | Eukaryotic translation initiation factor 5A-1 | 27 | −32.89 ± 3.17 | H205, D207, E381, R382, D410 | D1, S5, R16, K17, |
| DKVCVLSCGISTGLGASLNVAKP | P28032 | Alcohol dehydrogenase 2 | 23 | −32.30 ± 2.22 | V43, V184 | S11, L14 |
| DFLIGNTSTGYCAGG-CAAIV | Q40140 | Aspartic protease | 20 | −30.94 ± 2.68 | R31, H181, R187 *, E381 | D1, T9, A13 |
| Target: Staphylococcus aureus; UniProt: Q2FZP6; PDB: 4C12. * Residues of the active domain G110–S116, N151, T152–T513, S179, and R187. | ||||||
| DAGASKTYPQQAGTIRKGGHIVIKNRP | Q9AXQ6 | Eukaryotic translation initiation factor 5A-1 | 27 | −32.07 ± 4.96 | D5(A), E7(A), K8(A), R10(A), M1(B) | D1, A2, R16, G18, G19, H20 |
| Target: Helicobacter pylori; UniProt: O25608; PDB: 3QDL. * Residues of the active domain G150–G155. (A) and (B) denote the different chain. | ||||||
| DHVGFSCSTSGGAASRGILGPFGVIVIA | P29000 | Acid beta-fructofuranosidase | 28 | −49.54 ± 4.09 | T206(A), D63(B), E146(B), N148(B) | S8, T9, G12, R16, I18, G20 |
| DFLIGNTSTGYCAG-GCAAIV | Q40140 | Aspartic protease | 20 | −47.72 ± 1.56 | T206(A), A208(A), Y67(B), E146(B), G209(B), N210(B), V211(B), D213(B) | D1, S8, T9, Y11, A13, G14 |
| DAGASKTYPQQAGTIRKNGYIVIKGRP | Q9AXQ3 | Eukaryotic translation initiation factor 5A-4 | 27 | −45.81 ± 5.54 | D63(A) H116 * (A), D118 * (A), R205(A), N210(A), T206(B), A208(B) | D1, A2 G3, A4, K6, Y8, Q10, K17 |
| DKVCVLSCGISTGLGASLNVAKP | P28032 | Alcohol dehydrogenase 2 | 23 | −39.7 ± 2.04 | T206(A), D63(B), E146(B), N148(B) | C8, T12, S17, N19, K22 |
| DAGASKTYPQQAGTIRKGGHIVIKNRP | Q9AXQ6 | Eukaryotic translation initiation factor 5A-1 | 27 | −30.6 ± 1.28 | L203(A), D247(A), N254(A), D63(B), D117(B), D118 * (B), E146(B) | D1, K6, T7, K17, N25, R26 |
| Target: Pseudomonas aeruginosa; UniProt: D1MEN9; PDB: 6EW3. * Residues of the active domain H114, H116, D118, H153, H179, C198, and H240. (A) and (B) denote the different chains. | ||||||
| DHVGFSCSTSGGAA-SRGILGPFGVIVIA | P29000 | Acid beta-fructofuranosidase | 28 | −45.95 ± 3.77 | E87 *, D130 *, D178 *, H185, T189, R192, K205 | D1, H2, R16, I27 |
| DGPNASYITPAAL | P08196 | Phytoene synthase 1, chloroplastic | 13 | −44.92 ± 1.42 | D47 *, D49 *, E87 *, W112, R192, K205 | D1, S6 |
| DFLIGNTSTGYCAG-GCAAIV | Q40140 | Aspartic protease | 20 | −33.77 ± 3.73 | D47 *, E87, R105, Q126, W129, D130 *, D171, D182 | D1, L3, E5, N6, T7, Y11 |
| DKVCVLSCGISTGLGASLNVAKP | P28032 | Alcohol dehydrogenase 2 | 23 | −32.74 ± 5.71 | D128, D178, D182, H185, R192 | S11, L14, G15, L18, N19 |
| Target: Salmonella enterica subsp. enterica serovar Typhimurium str. LT2; UniProt: Q8ZPX9, PDB: 6FZB. * Residues of the active domain S36, S46, D47, D49, E87, D130, W173–D178, H185, K205, and Y231. | ||||||
| DAGASKTYPQQAGTIRKGGHIVIKNRP | Q9AXQ6 | Eukaryotic translation initiation factor 5A-1 | 27 | −32.07 ± 1.49 | D311, E676 *, G678 *, T679 *, I742, N743 | S5, T7, Y8, K24, R26 |
| DKVCVLSCGISTGLGASLNVAKP | P28032 | Alcohol dehydrogenase 2 | 23 | −31.44 ± 2.67 | Y180 | T12, N19 |
| Target: Acinetobacter baumannii; UniProt: G1C794, PDB: 3UDF. * Residues of the active domain G321–A737. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morena, F.; Cencini, C.; Calzoni, E.; Martino, S.; Emiliani, C. A Novel Workflow for In Silico Prediction of Bioactive Peptides: An Exploration of Solanum lycopersicum By-Products. Biomolecules 2024, 14, 930. https://doi.org/10.3390/biom14080930
Morena F, Cencini C, Calzoni E, Martino S, Emiliani C. A Novel Workflow for In Silico Prediction of Bioactive Peptides: An Exploration of Solanum lycopersicum By-Products. Biomolecules. 2024; 14(8):930. https://doi.org/10.3390/biom14080930
Chicago/Turabian StyleMorena, Francesco, Chiara Cencini, Eleonora Calzoni, Sabata Martino, and Carla Emiliani. 2024. "A Novel Workflow for In Silico Prediction of Bioactive Peptides: An Exploration of Solanum lycopersicum By-Products" Biomolecules 14, no. 8: 930. https://doi.org/10.3390/biom14080930