
Harnessing Porphyrin Accumulation in Liver Cancer: Combining Genomic Data and Drug Targeting


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Primary Liver Cell and Hepatoma Cell Line Preparation
2.2. Bait-and-Kill Drug Assay
2.3. RNAseq Experiments of Primary Human Hepatocytes and Cell Lines
2.4. scRNAseq of Primary Human Hepatic Cell Populations
2.5. HCC Clinical Progression Patterns via Transcriptome Data Analysis
2.6. Patient Tumor Gene Expression Analysis and Survival Analysis
3. Results
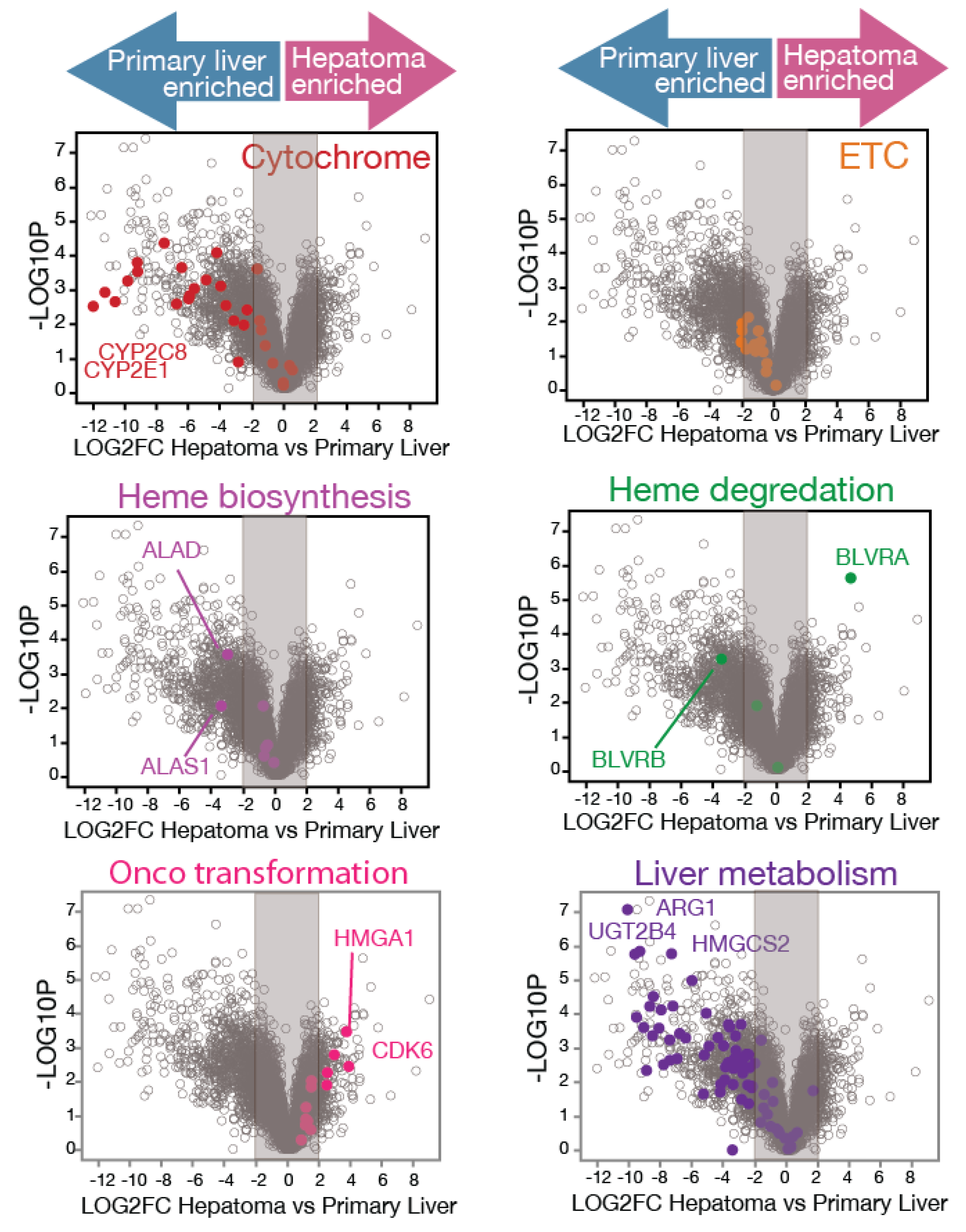
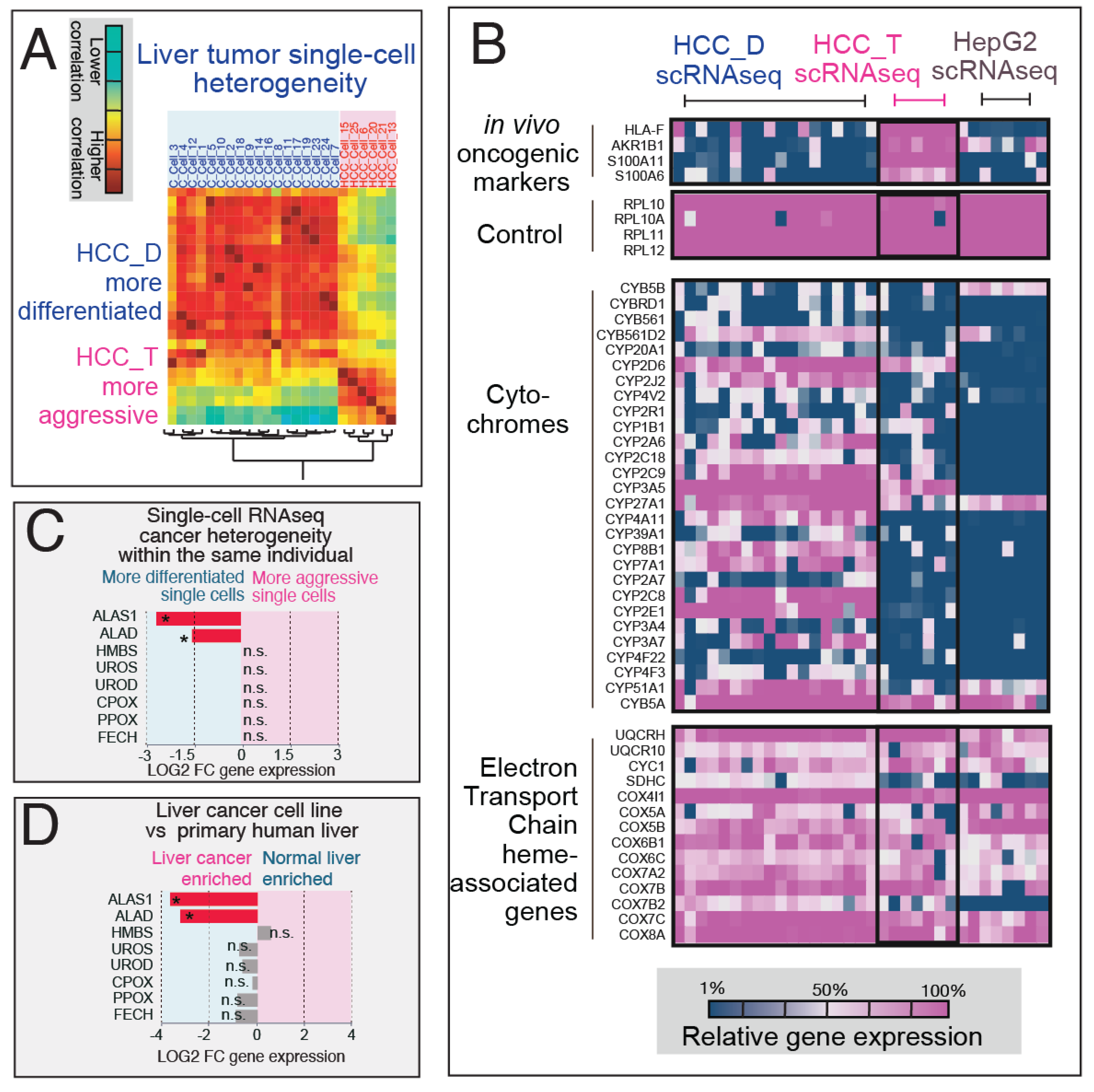
3.1. RNAseq Analysis Reveals Mass Alterations in Heme Metabolism Between Normal Primary Human Hepatocytes and Hepatoma Cell Lines
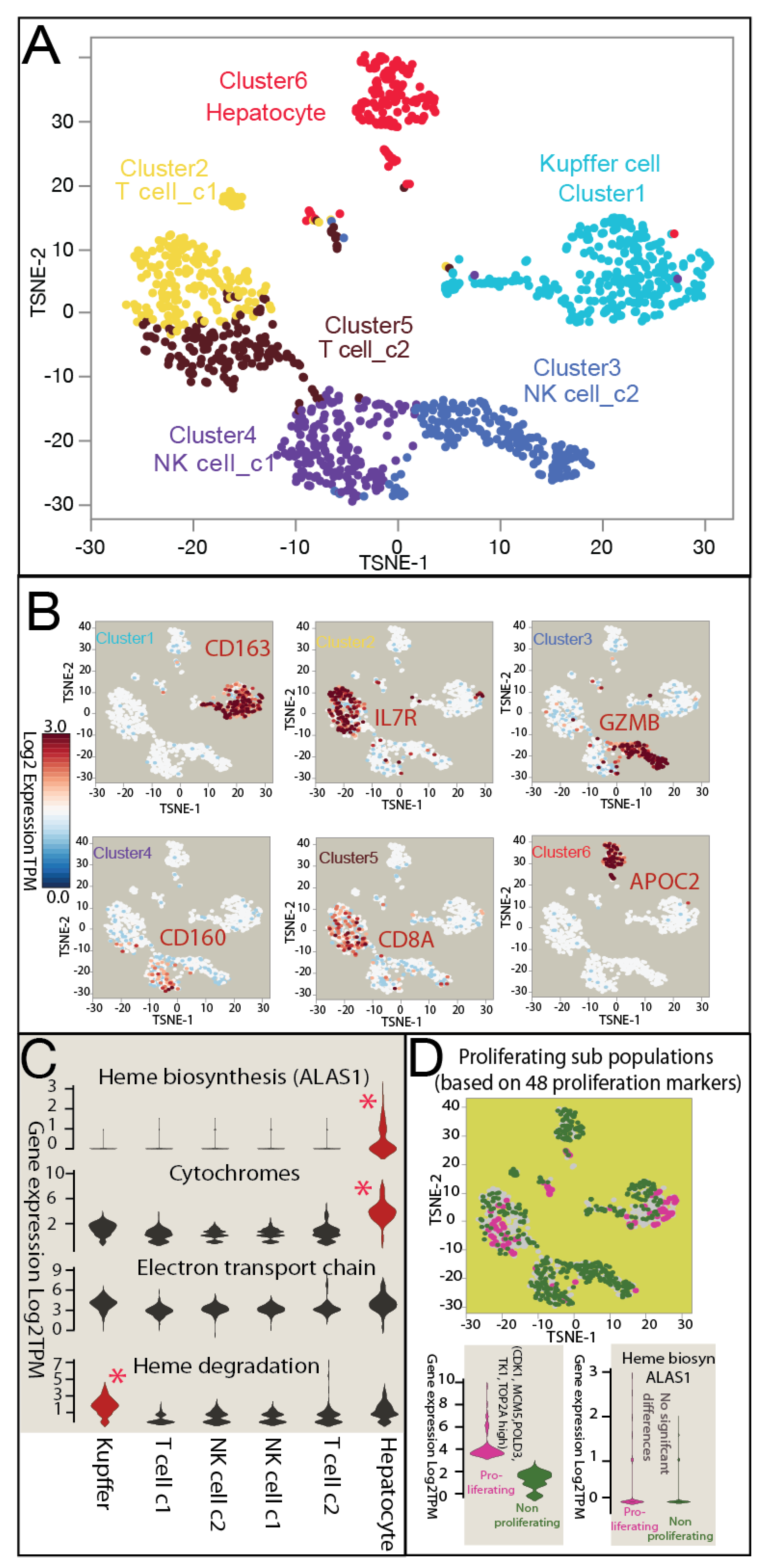
3.2. Single-Cell Transcriptome Analysis Reveals Heme Rewiring Associated with Cell-to-Cell Heterogeneity in Oncogenesis in Human Liver Cancer
3.3. Heterogeneous Distribution of Heme Metabolism across Normal Human Liver Cell Populations in scRNAseq
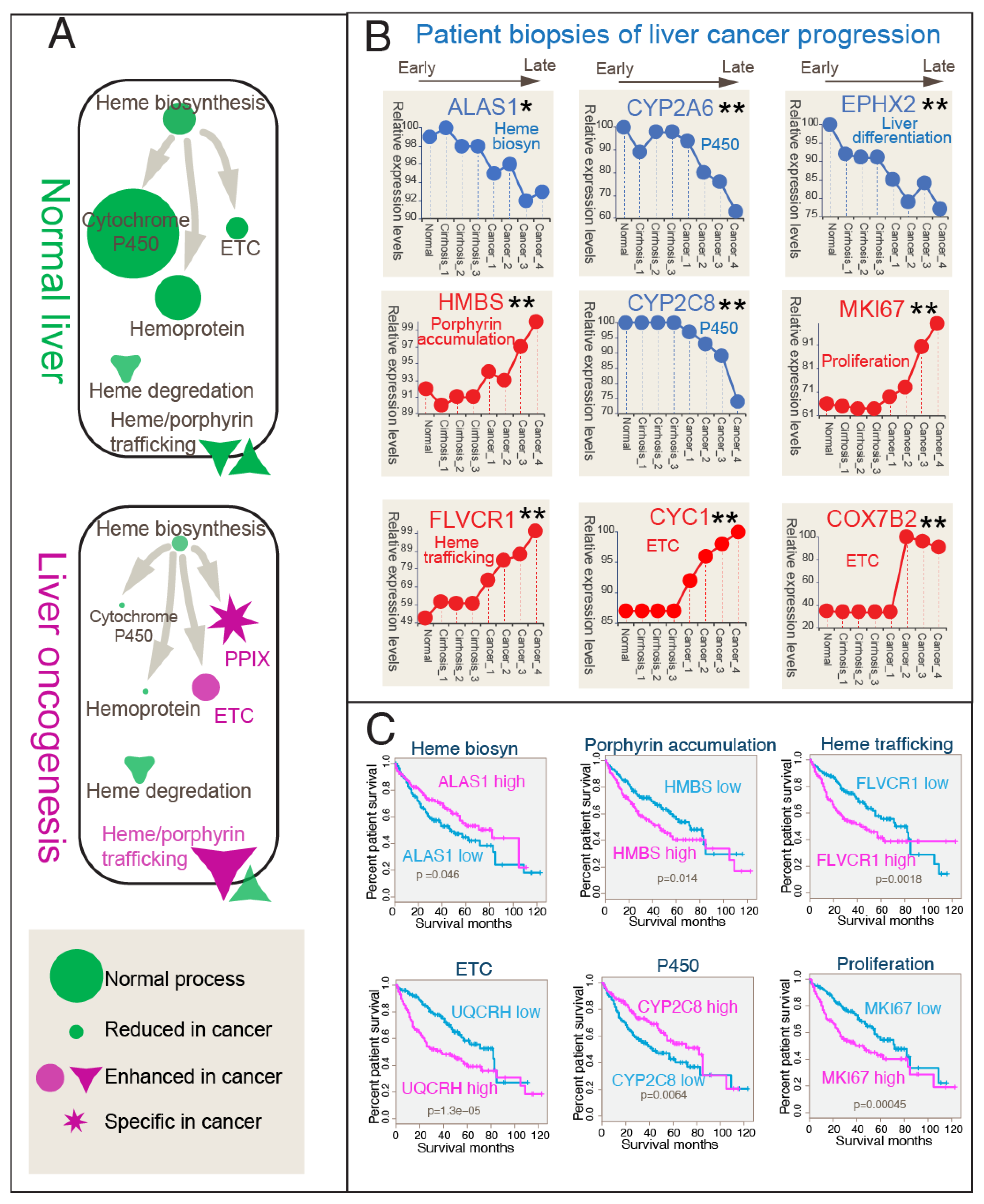
3.4. Correlation Between Heme Metabolic Shift and Clinical Progression in Liver Cancer Patient Gene Expressions
3.5. Targeting Liver Cancer Porphyrin Overdrive In Vitro with a Bait-and-Kill Strategy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arias, I.; Jakoby, W.; Popper, M.; Schachter, D. The Liver-Biology and Pathobiology; Shafritz Raven Press: New York, NY, USA, 1988. [Google Scholar]
- Balwani, M.; Wang, B.; Anderson, K.E.; Bloomer, J.R.; Bissell, D.M.; Bonkovsky, H.L.; Phillips, J.D.; Desnick, R.J.; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Acute hepatic porphyrias: Recommendations for evaluation and long-term management. Hepatology 2017, 66, 1314–1322. [Google Scholar] [CrossRef]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Caldwell, S.H.; Intagliata, N.M. Haemostatic alterations and management of haemostasis in patients with cirrhosis. J. Hepatol. 2022, 76, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Vimalesvaran, S.; Dhawan, A. Liver transplantation for pediatric inherited metabolic liver diseases. World J. Hepatol. 2021, 13, 1351–1366. [Google Scholar] [CrossRef]
- Członkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome P450s and other enzymes in drug metabolism and toxicity. AAPS J. 2006, 8, E101–E111. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Guengerich, F.P. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 2015, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Rojas Velazquez, M.N.; Therkelsen, S.; Pandey, A.V. Exploring novel variants of the cytochrome p450 reductase gene (por) from the genome aggregation database by integrating bioinformatic tools and functional assays. Biomolecules 2023, 13, 1728. [Google Scholar] [CrossRef]
- Lee, J.; Beers, J.L.; Geffert, R.M.; Jackson, K.D. A review of cyp-mediated drug interactions: Mechanisms and in vitro drug-drug interaction assessment. Biomolecules 2024, 14, 99. [Google Scholar] [CrossRef]
- Esteves, F.; Almeida, C.M.M.; Silva, S.; Saldanha, I.; Urban, P.; Rueff, J.; Pompon, D.; Truan, G.; Kranendonk, M. Single mutations in cytochrome p450 oxidoreductase can alter the specificity of human cytochrome p450 1a2-mediated caffeine metabolism. Biomolecules 2023, 13, 1083. [Google Scholar] [CrossRef] [PubMed]
- Fronczek, M.; Strzelczyk, J.K.; Biernacki, K.; Salatino, S.; Osadnik, T.; Ostrowska, Z. New variants of the cytochrome p450 2r1 (cyp2r1) gene in individuals with severe vitamin d-activating enzyme 25(oh)d deficiency. Biomolecules 2021, 11, 1867. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.R.; Dangi, B.; Yue, G.; Ahire, D.S.; Prasad, B.; Zgoda, V.G. Exploring the interactome of cytochrome p450 2e1 in human liver microsomes with chemical crosslinking mass spectrometry. Biomolecules 2022, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Idda, M.L.; Campesi, I.; Fiorito, G.; Vecchietti, A.; Urru, S.A.M.; Solinas, M.G.; Franconi, F.; Floris, M. Sex-biased expression of pharmacogenes across human tissues. Biomolecules 2021, 11, 1206. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. A new cytochrome in liver microsomes. J. Biol. Chem. 1962, 237, 1375–1376. [Google Scholar] [CrossRef]
- De Montellano, P.R.O. Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Daly, A. Pharmacogenetics of the major polymorphic metabolizing enzymes. Fundam. Clin. Pharmacol. 2003, 17, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Turilli-Ghisolfi, E.S.; Lualdi, M.; Fasano, M. Ligand-based regulation of dynamics and reactivity of hemoproteins. Biomolecules 2023, 13, 683. [Google Scholar] [CrossRef]
- Xu, R.-H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Kalainayakan, S.P.; FitzGerald, K.E.; Konduri, P.C.; Vidal, C.; Zhang, L. Essential roles of mitochondrial and heme function in lung cancer bioenergetics and tumorigenesis. Cell Biosci. 2018, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Segaran, A.; Lord, S. Targeting OXPHOS and the electron transport chain in cancer; Molecular and therapeutic implications. Semin Cancer Biol 2022, 86, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Bedi, M.; Ray, M.; Ghosh, A. Active mitochondrial respiration in cancer: A target for the drug. Mol. Cell. Biochem. 2022, 477, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Adapa, S.R.; Hunter, G.A.; Amin, N.E.; Marinescu, C.; Borsky, A.; Sagatys, E.M.; Sebti, S.M.; Reuther, G.W.; Ferreira, G.C.; Jiang, R.H. Porphyrin overdrive rewires cancer cell metabolism. Life Sci. Alliance 2024, 7, e202302547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Heme Biology: The Secret Life of Heme in Regulating Diverse Biological Processes; World Scientific: Singapore, 2011. [Google Scholar]
- Barace, S.; Santamaría, E.; Infante, S.; Arcelus, S.; De La Fuente, J.; Goñi, E.; Tamayo, I.; Ochoa, I.; Sogbe, M.; Sangro, B.; et al. Application of graph models to the identification of transcriptomic oncometabolic pathways in human hepatocellular carcinoma. Biomolecules 2024, 14, 653. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, O.N.; Krasnov, G.S.; Snezhkina, A.V.; Kudryavtseva, A.V.; Fedorov, V.S.; Zakirova, N.F.; Golikov, M.V.; Kochetkov, S.N.; Bartosch, B.; Valuev-Elliston, V.T.; et al. Transcriptome analysis of redox systems and polyamine metabolic pathway in hepatoma and non-tumor hepatocyte-like cells. Biomolecules 2023, 13, 714. [Google Scholar] [CrossRef] [PubMed]
- Morse, P.T.; Wan, J.; Arroum, T.; Herroon, M.K.; Kalpage, H.A.; Bazylianska, V.; Lee, I.; Heath, E.I.; Podgorski, I.; Hüttemann, M. Prostate cancer-specific lysine 53 acetylation of cytochrome c drives metabolic reprogramming and protects from apoptosis in intact cells. Biomolecules 2024, 14, 695. [Google Scholar] [CrossRef]
- New-Aaron, M.; Thomes, P.G.; Ganesan, M.; Dagur, R.S.; Donohue, T.M.; Kusum, K.K.; Poluektova, L.Y.; Osna, N.A. Alcohol-induced lysosomal damage and suppression of lysosome biogenesis contribute to hepatotoxicity in hiv-exposed liver cells. Biomolecules 2021, 11, 1497. [Google Scholar] [CrossRef]
- Fiorito, V.; Allocco, A.L.; Petrillo, S.; Gazzano, E.; Torretta, S.; Marchi, S.; Destefanis, F.; Pacelli, C.; Audrito, V.; Provero, P.; et al. The heme synthesis-export system regulates the tricarboxylic acid cycle flux and oxidative phosphorylation. Cell Rep. 2021, 35, 109252. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Nagar, S.; Bhagwat, M.; Uddin, M.; Zhu, Y.; Vancurova, I.; Vancura, A. Activated heme synthesis regulates glycolysis and oxidative metabolism in breast and ovarian cancer cells. PLoS ONE 2021, 16, e0260400. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Wurmbach, E.; Chen, Y.B.; Khitrov, G.; Zhang, W.; Roayaie, S.; Schwartz, M.; Fiel, I.; Thung, S.; Mazzaferro, V.; Bruix, J.; et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007, 45, 938–947. [Google Scholar] [CrossRef]
- Chang, K.; Creighton, C.J.; Davis, C.; Donehower, L.; Drummond, J.; Wheeler, D.; Ally, A.; Balasundaram, M.; Birol, I.; Butterfield, Y.S.N.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Ardlie, K. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue 1252 gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar]
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Mantel, N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar] [PubMed]
- Maher, S.P.; Conway, A.J.; Roth, A.; Adapa, S.R.; Cualing, P.; Andolina, C.; Hsiao, J.; Turgeon, J.; Chaumeau, V.; Johnson, M.; et al. An adaptable soft-mold embossing process for fabricating optically-accessible, microfeature-based culture systems and application toward liver stage antimalarial compound testing. Lab Chip 2020, 20, 1124–1139. [Google Scholar] [CrossRef]
- Hunter, G.A.; Ferreira, G.C. Molecular enzymology of 5-Aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta 2011, 1814, 1467–1473. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, A.; Quan, C.; Pan, Y.; Zhang, H.; Li, Y.; Gao, C.; Lu, H.; Wang, X.; Cao, P.; et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat. Commun. 2022, 13, 4594. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of infiltrating t cells in liver cancer revealed by single-cell sequencing. Cell 2017, 169, 1342–1356.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, M.; Ling, B.; Liu, S.; Zheng, Y.; Nie, C.; Yuan, Z.; Zhou, L.; Guo, G.; Tong, A.; et al. Increased expression of S100A6 promotes cell proliferation and migration in human hepatocellular carcinoma. J. Mol. Med. 2014, 92, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, W.; Lu, J.; Liu, Y.; Zhou, W.; Yu, Z.; Qin, S.; Fan, J. HCC: RNA-sequencing in cirrhosis. Biomolecules 2023, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Rizzardini, M.; Zappone, M.; Villa, P.; Gnocchi, P.; Sironi, M.; Diomede, L.; Meazza, C.; Monshouwer, M.; Cantoni, L. Kupffer cell depletion partially prevents hepatic heme oxygenase 1 messenger RNA accumulation in systemic inflammation in mice: Role of interleukin 1beta. Hepatology 1998, 27, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Chen, S.; Chen, X.; Zhu, C.; Tang, C.; Wang, S.; Gao, Y.; Fu, S.; Liu, Q. Integrating multiple references for single-cell assignment. Nucleic Acids Res. 2021, 49, e80. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef] [PubMed]
- Zhan, K.; Bai, Y.; Liao, S.; Chen, H.; Kuang, L.; Luo, Q.; Lv, L.; Qiu, L.; Mei, Z. Identification and validation of EPHX2 as a prognostic biomarker in hepatocellular carcinoma. Mol. Med. Rep. 2021, 24, 650. [Google Scholar] [CrossRef] [PubMed]
- Adapa, S.R.; Sami, A.; Meshram, P.; Ferreira, G.C.; Jiang, R.H.Y. Uncovering porphyrin accumulation in the tumor microenvironment. Genes 2024, 15, 961. [Google Scholar] [CrossRef]
- Ozten, O.; Guney Eskiler, G.; Sonmez, F.; Yıldız, M.Z. Investigation of the therapeutic effect of 5-aminolevulinic acid based photodynamic therapy on hepatocellular carcinoma. Lasers Med. Sci. 2022, 37, 1325–1332. [Google Scholar] [CrossRef]
- Nanashima, A.; Hiyoshi, M.; Imamura, N.; Yano, K.; Hamada, T.; Kai, K. Recent Advances in Photodynamic Imaging and Therapy in Hepatobiliary Malignancies: Clinical and Experimental Aspects. Curr. Oncol. 2021, 28, 4067–4079. [Google Scholar] [CrossRef]
- Svanberg, K.; Liu, D.L.; Wang, I.; Andersson-Engels, S.; Stenram, U.; Svanberg, S. Photodynamic therapy using intravenous delta-aminolaevulinic acid-induced protoporphyrin IX sensitisation in experimental hepatic tumours in rats. Br. J. Cancer 1996, 74, 1526–1533. [Google Scholar] [CrossRef]
- Otake, M.; Nishiwaki, M.; Kobayashi, Y.; Baba, S.; Kohno, E.; Kawasaki, T.; Fujise, Y.; Nakamura, H. Selective accumulation of ALA-induced PpIX and photodynamic effect in chemically induced hepatocellular carcinoma. Br. J. Cancer 2003, 89, 730–736. [Google Scholar] [CrossRef]
- Szeimies, R.M.; Abels, C.; Fritsch, C.; Karrer, S.; Steinbach, P.; Bäumler, W.; Goerz, G.; Goetz, A.E.; Landthaler, M. Wavelength dependency of photodynamic effects after sensitization with 5-aminolevulinic acid in vitro and in vivo. J. Investig. Dermatol. 1995, 105, 672–677. [Google Scholar] [CrossRef]
- Kaneko, S.; Kaneko, S. Fluorescence-Guided Resection of Malignant Glioma with 5-ALA. Int. J. Biomed. Imaging 2016, 2016, 6135293. [Google Scholar] [CrossRef]
- Moesta, K.T.; Ebert, B.; Handke, T.; Nolte, D.; Nowak, C.; Haensch, W.E.; Pandey, R.K.; Dougherty, T.J.; Rinneberg, H.; Schlag, P.M. Protoporphyrin IX occurs naturally in colorectal cancers and their metastases. Cancer Res. 2001, 61, 991–999. [Google Scholar]
- Bellini, M.H.; Coutinho, E.L.; Courrol, L.C.; Rodrigues de Oliveira Silva, F.; Vieira Júnior, N.D.; Schor, N. Correlation between autofluorescence intensity and tumor area in mice bearing renal cell carcinoma. J. Fluoresc. 2008, 18, 1163–1168. [Google Scholar] [CrossRef]
- Yanatori, I.; Yasui, Y.; Miura, K.; Kishi, F. Mutations of FLVCR1 in posterior column ataxia and retinitis pigmentosa result in the loss of heme export activity. Blood Cells Mol. Dis. 2012, 49, 60–66. [Google Scholar] [CrossRef]
- Mukherjee, A.; Ghosh, K.K.; Chakrabortty, S.; Gulyás, B.; Padmanabhan, P.; Ball, W.B. Mitochondrial reactive oxygen species in infection and immunity. Biomolecules 2024, 14, 670. [Google Scholar] [CrossRef]
- Lynch, J.; Wang, Y.; Li, Y.; Kavdia, K.; Fukuda, Y.; Ranjit, S.; Robinson, C.G.; Grace, C.R.; Xia, Y.; Peng, J.; et al. A PPIX-binding probe facilitates discovery of PPIX-induced cell death modulation by peroxiredoxin. Commun. Biol. 2023, 6, 673. [Google Scholar] [CrossRef]
- Pignatelli, P.; Umme, S.; D’Antonio, D.L.; Piattelli, A.; Curia, M.C. Reactive Oxygen Species Produced by 5-Aminolevulinic Acid Photodynamic Therapy in the Treatment of Cancer. Int. J. Mol. Sci. 2023, 24, 8964. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, C.-J.; Chia, W.N.; Loh, C.C.Y.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.-X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.N.; Damle, S.S.; Moore, E.E.; Ao, L.; Song, Y.; Johnson, J.L.; Weyant, M.; Banerjee, A.; Meng, X.; Fullerton, D.A. Hemoglobin-based oxygen carrier induces hepatic heme oxygenase 1 expression in Kupffer cells. Surgery 2007, 142, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Hirano, K.; Yamamoto, T.; Hasegawa, G.; Hatakeyama, K.; Suematsu, M.; Naito, M. The protective role of Kupffer cells in the ischemia-reperfused rat liver. Arch. Histol. Cytol. 2002, 65, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.A.; McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Schmidt, C.; Kassai, M.; Kew, K.A.; McClung, J.M.; Neufer, P.D.; Brophy, P.; et al. Intrinsic OXPHOS limitations underlie cellular bioenergetics in leukemia. eLife 2021, 10, e63104. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Finley, L.W.S. What is cancer metabolism? Cell 2023, 186, 1670–1688. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W. The significance of mitochondrial dysfunction in cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef]
- Voltarelli, V.A.; Alves de Souza, R.W.; Miyauchi, K.; Hauser, C.J.; Otterbein, L.E. Heme: The lord of the iron ring. Antioxidants 2023, 12, 1074. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adapa, S.R.; Meshram, P.; Sami, A.; Jiang, R.H.Y. Harnessing Porphyrin Accumulation in Liver Cancer: Combining Genomic Data and Drug Targeting. Biomolecules 2024, 14, 959. https://doi.org/10.3390/biom14080959
Adapa SR, Meshram P, Sami A, Jiang RHY. Harnessing Porphyrin Accumulation in Liver Cancer: Combining Genomic Data and Drug Targeting. Biomolecules. 2024; 14(8):959. https://doi.org/10.3390/biom14080959
Chicago/Turabian StyleAdapa, Swamy R., Pravin Meshram, Abdus Sami, and Rays H. Y. Jiang. 2024. "Harnessing Porphyrin Accumulation in Liver Cancer: Combining Genomic Data and Drug Targeting" Biomolecules 14, no. 8: 959. https://doi.org/10.3390/biom14080959
APA StyleAdapa, S. R., Meshram, P., Sami, A., & Jiang, R. H. Y. (2024). Harnessing Porphyrin Accumulation in Liver Cancer: Combining Genomic Data and Drug Targeting. Biomolecules, 14(8), 959. https://doi.org/10.3390/biom14080959