

Mechanism of Reactive Oxygen Species-Guided Immune Responses in Gouty Arthritis and Potential Therapeutic Targets

Abstract
:1. Introduction
2. Mechanisms of GA Onset and Its Link to ROS
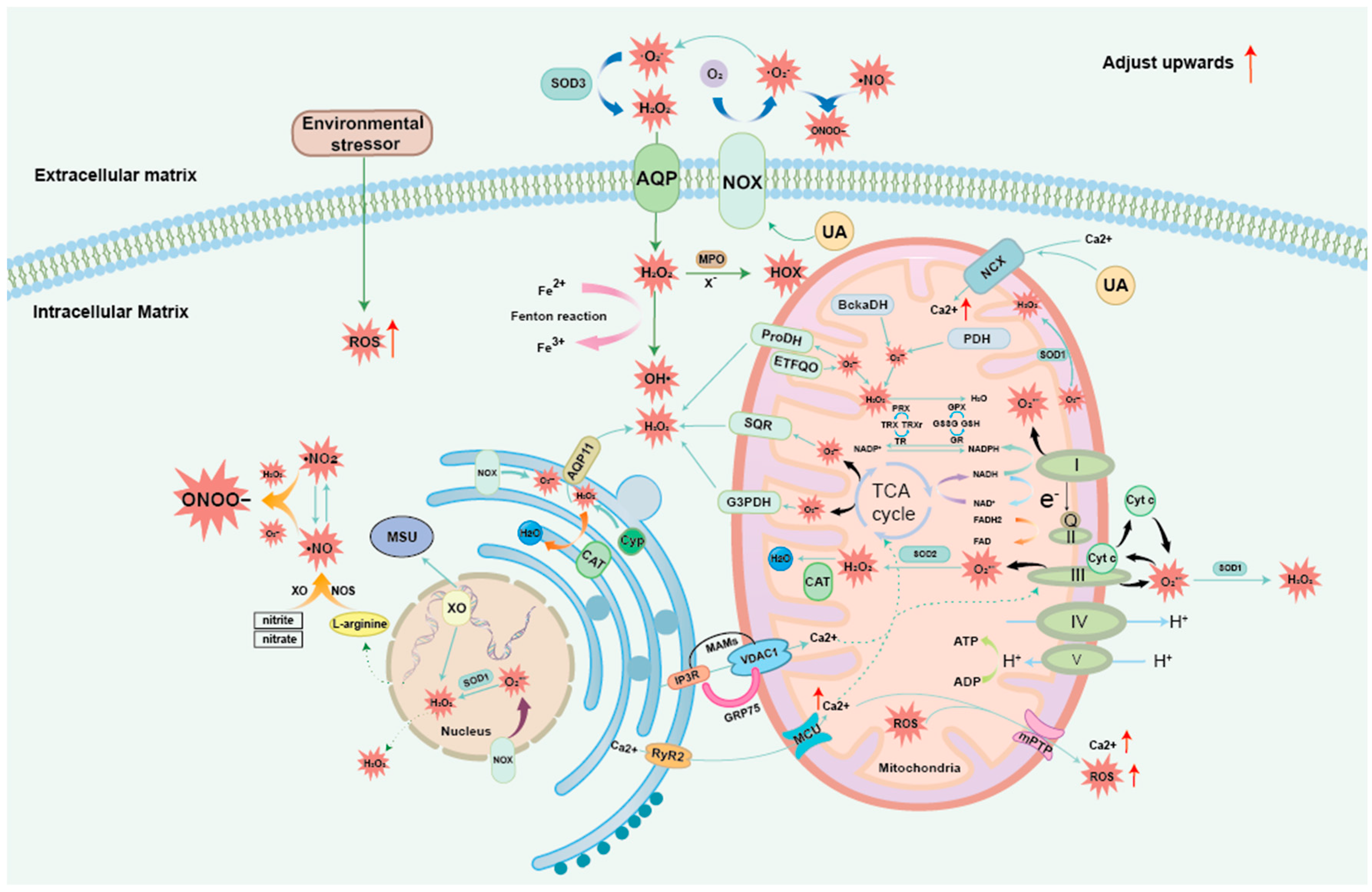
3. ROS and OS Generation Mechanism
3.1. Exogenous ROS
3.2. Mitochondrial ROS
3.3. Common ROS: H2O2
3.4. Nitrogen Oxides
3.5. Endoplasmic Reticulum-Mitochondria ROS
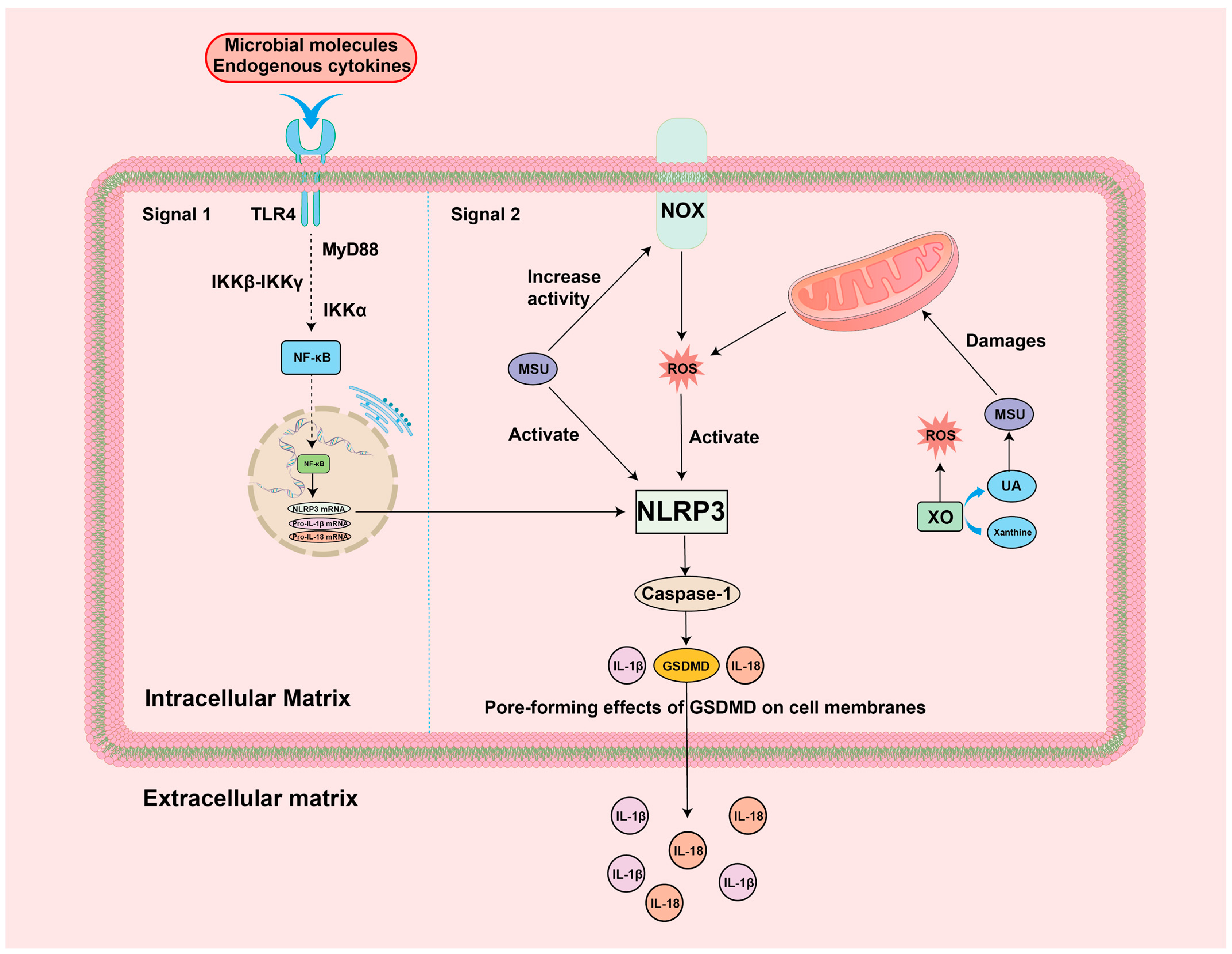
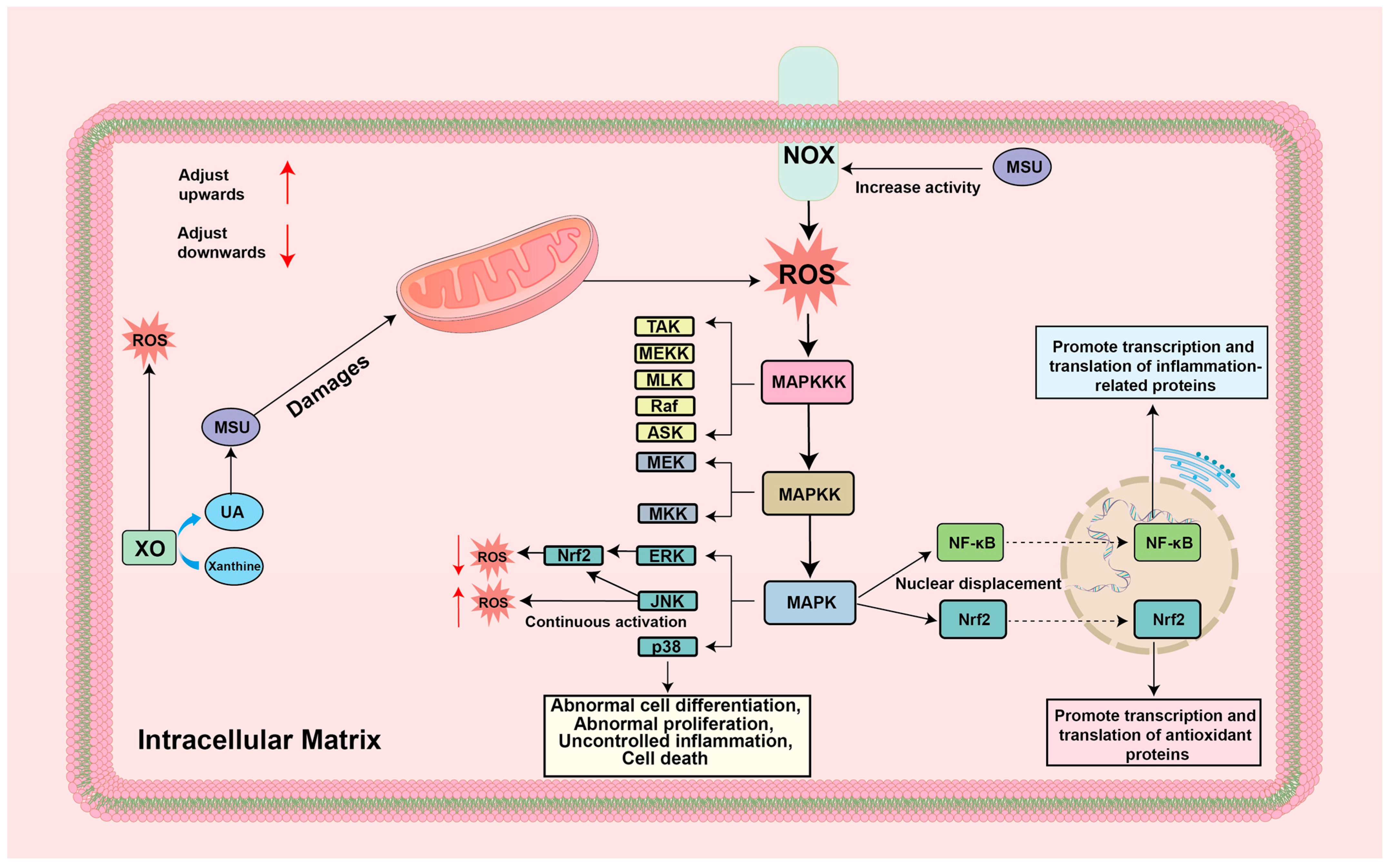
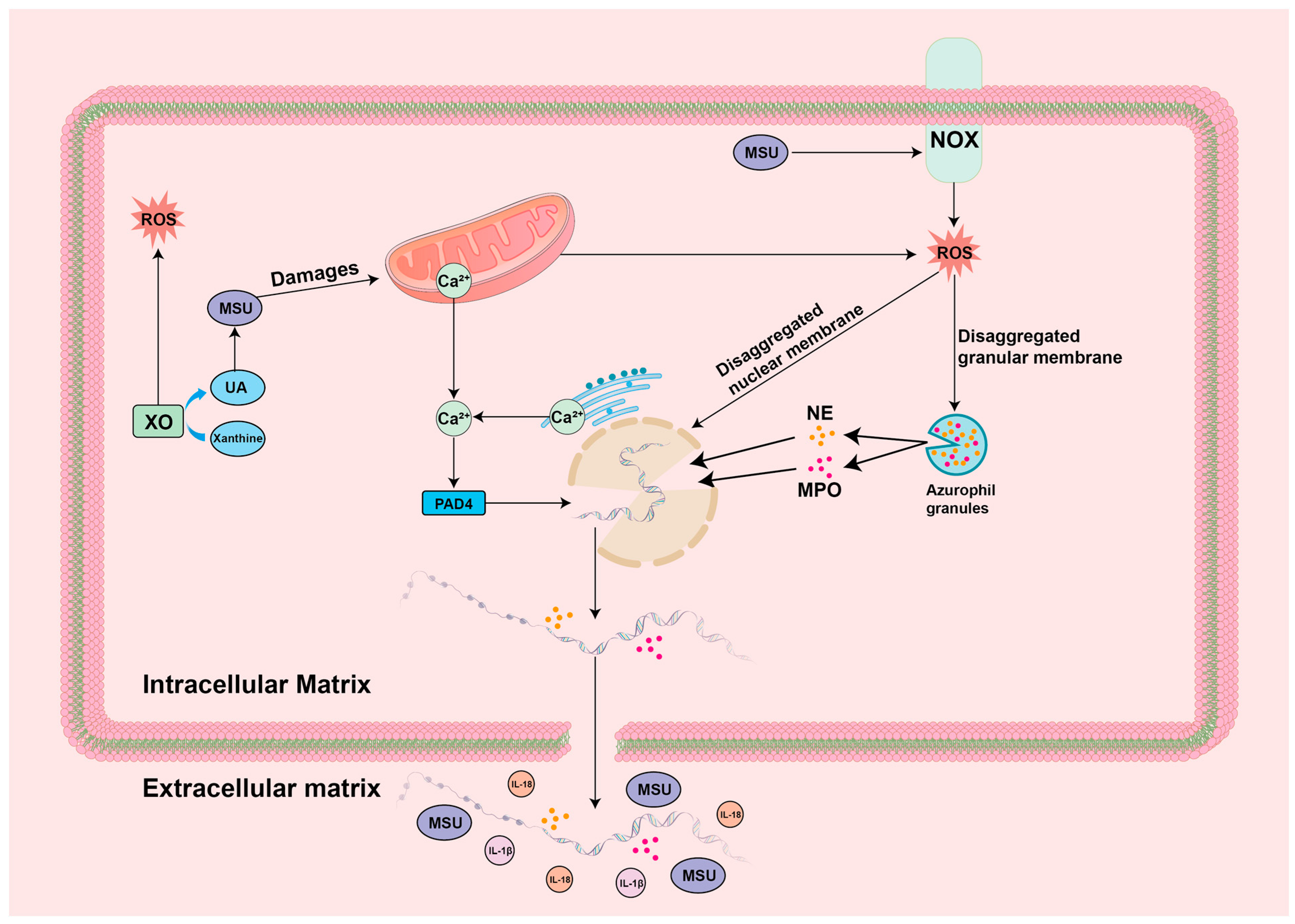
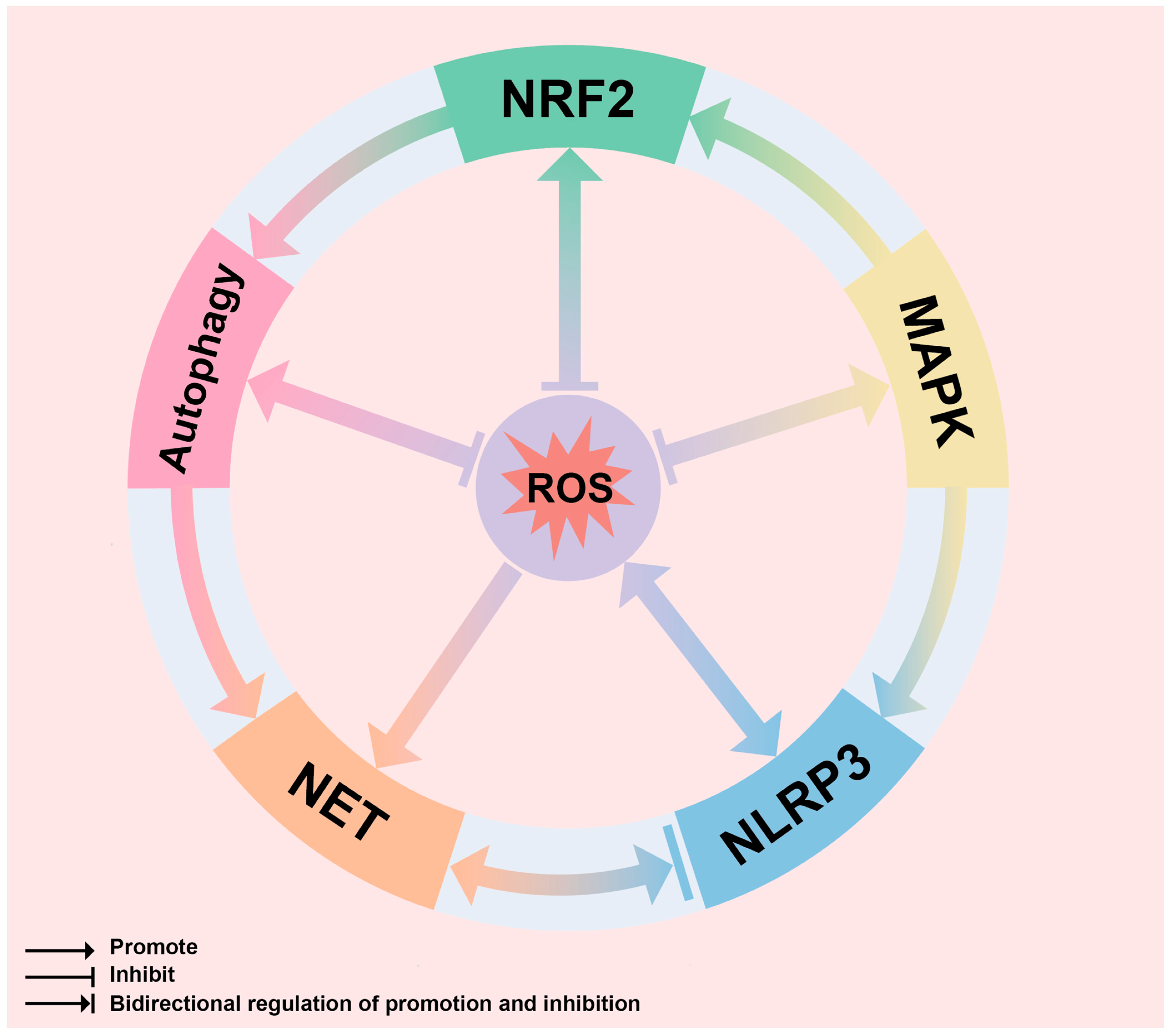
4. Mechanisms of ROS-Guided Immune Responses in the GA Environment
4.1. ROS-NF-κB-NLRP3 Inflammation
4.2. ROS-MAPK
4.3. ROS-NET
4.4. ROS-Autophagy
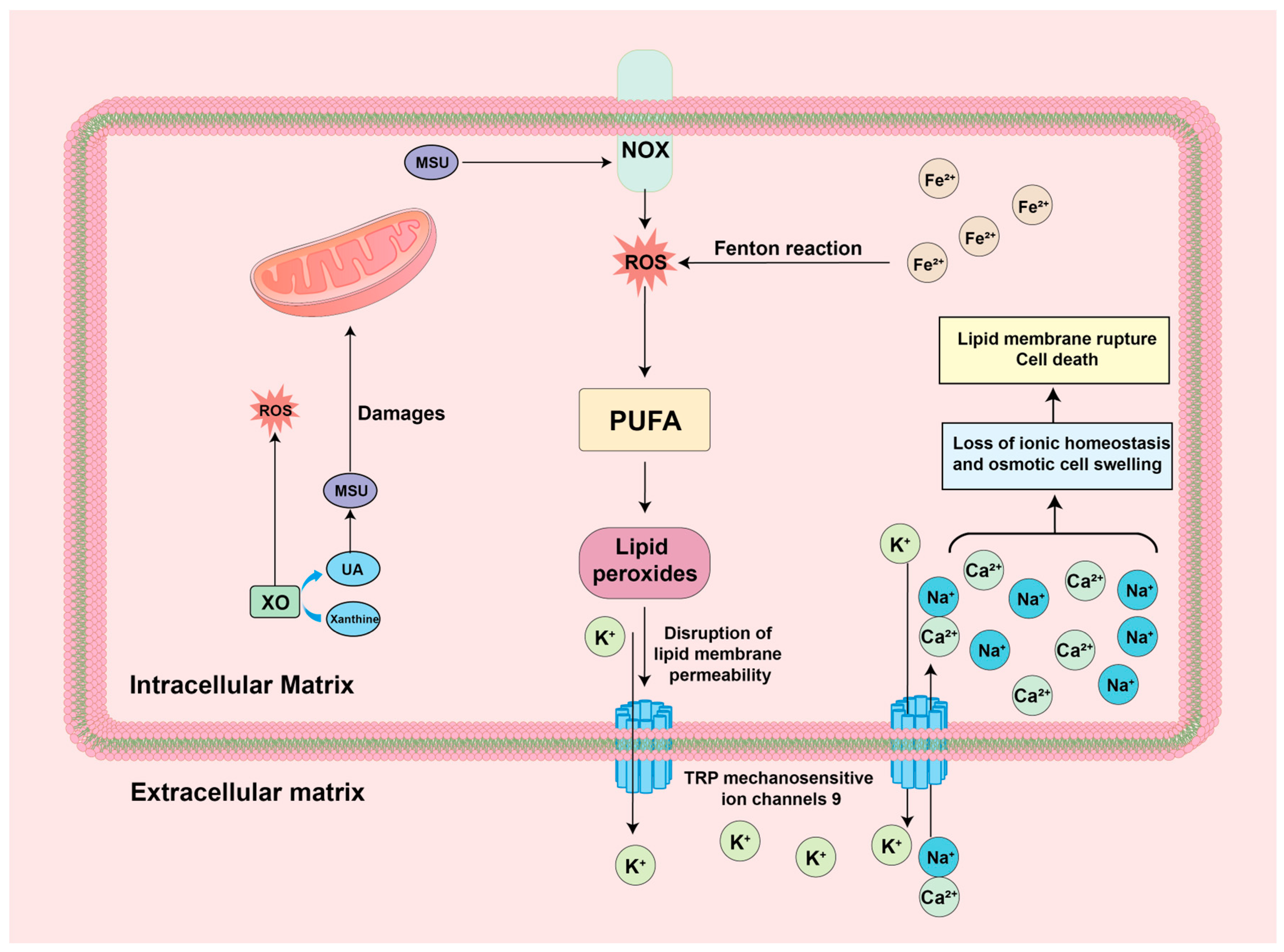
4.5. ROS-Ferroptosis
4.6. ROS-Nrf2 Antioxidant Response
5. Targeted ROS Network Drug Therapy for GA
5.1. Targeting ROS-NLRP3 for the Treatment of GA
5.2. Targeting ROS-MAPK for the Treatment of GA
5.3. Targeting ROS-NET for the Treatment of GA
5.4. Targeting ROS-Autophagy for the Treatment of GA
5.5. Targeting ROS-Ferroptosis for the Treatment of GA
5.6. Targeting ROS-Nrf2 for the Treatment of GA
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Arakawa, H.; Tamai, I. Uric acid in health and disease: From physiological functions to pathogenic mechanisms. Pharmacol. Ther. 2024, 256, 108615. [Google Scholar] [CrossRef] [PubMed]
- Neogi, T. Clinical practice. Gout. N. Engl. J. Med. 2011, 364, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Li, X.; Xie, B.; Lai, Y.; Sosnik, A.; Boucetta, H.; Chen, Z.; He, W. Gout therapeutics and drug delivery. J. Control Release 2023, 362, 728–754. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-R.; Wang, J.-Q.; Li, J. Role of NLRP3 in the pathogenesis and treatment of gout arthritis. Front. Immunol. 2023, 14, 1137822. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, H.S.; Yang, G.; Kim, H.J. Natural Products as a Novel Therapeutic Strategy for NLRP3 Inflammasome-Mediated Gout. Front. Pharmacol. 2022, 13, 861399. [Google Scholar] [CrossRef] [PubMed]
- Stamp, L.K.; Turner, R.; Khalilova, I.S.; Zhang, M.; Drake, J.; Forbes, L.V.; Kettle, A.J. Myeloperoxidase and oxidation of uric acid in gout: Implications for the clinical consequences of hyperuricaemia. Rheumatology 2014, 53, 1958–1965. [Google Scholar] [CrossRef]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Alduraibi, F.K.; Saleem, M.; Ricart, K.; Patel, R.P.; Szalai, A.J.; Singh, J.A. Clustering Patients With Gout Based on Comorbidities and Biomarkers: A Cross-Sectional Study. J. Rheumatol. 2023, 50, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Hasikova, L.; Kozlik, P.; Kalikova, K.; Stiburkova, B.; Zavada, J. OP0206 allantoin—A biomarker of oxidative stress—Is higher in patients with gout than in healthy volunteers, and corresponds with severity of disease. Ann. Rheum. Dis. 2020, 79 (Suppl. S1), 128. [Google Scholar] [CrossRef]
- Khalfina, T.; Maksudova, A. AB0849 Oxidative Stress, Anti-Oxidant Activity in Patients with Gout. Ann. Rheum. Dis. 2014, 73 (Suppl. S2), 1083. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, M.; Pu, Z.; Xu, G.; Li, X. Relationship between oxidative stress and inflammation in hyperuricemia: Analysis based on asymptomatic young patients with primary hyperuricemia. Medicine 2018, 97, e13108. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, S.; Elsaid, K.A. Protein phosphatase 2A regulates xanthine oxidase-derived ROS production in macrophages and influx of inflammatory monocytes in a murine gout model. Front. Pharmacol. 2022, 13, 1033520. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, L.; Rudin, A.D.; Klose, F.P.S.; Buck, A.; Björkman, L.; Christenson, K.; Bylund, J. In Vivo Transmigrated Human Neutrophils Are Highly Primed for Intracellular Radical Production Induced by Monosodium Urate Crystals. Int. J. Mol. Sci. 2020, 21, 3750. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, H.; Sun, Q.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. The Role of Oxidative Stress in Hyperuricemia and Xanthine Oxidoreductase (XOR) Inhibitors. Oxidative Med. Cell. Longev. 2021, 2021, 1470380. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, R.; Pasalar, P.; Shokri, H.; Shabani, M.; Emamgholipour, S. Evidence for the effect of soluble uric acid in augmenting endoplasmic reticulum stress markers in human peripheral blood mononuclear cells. J. Physiol. Biochem. 2022, 78, 343–353. [Google Scholar] [CrossRef]
- Zhang, B.; Di, H.; Zhang, Y.; Han, X.; Yin, Y.; Han, Y.; Cao, Y.; Zeng, X. Time- and Concentration-Dependent Stimulation of Oxidative Stress in Chondrocytes by Intracellular Soluble Urate. Curr. Mol. Med. 2024, 24, 233–243. [Google Scholar] [CrossRef]
- He, B.; Nie, Q.; Wang, F.; Wang, X.; Zhou, Y.; Wang, C.; Guo, J.; Fan, X.; Ye, Z.; Liu, P.; et al. Hyperuricemia promotes the progression of atherosclerosis by activating endothelial cell pyroptosis via the ROS/NLRP3 pathway. J. Cell. Physiol. 2023, 238, 1808–1822. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, J.; Liang, Z.; Li, M.; Fu, D.; Zhang, L.; Yang, X.; Guo, Y.; Ge, D.; Liu, Y.; et al. Monosodium urate crystals with controlled shape and aspect ratio for elucidating the pathological progress of acute gout. Biomater. Adv. 2022, 139, 213005. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Cuevas, Y.; Martínez-Flores, K.; Fernández-Torres, J.; Loissell-Baltazar, Y.A.; Medina-Luna, D.; López-Macay, A.; Camacho-Galindo, J.; Hernández-Díaz, C.; Santamaría-Olmedo, M.G.; López-Villegas, E.O.; et al. Monosodium urate crystals induce oxidative stress in human synoviocytes. Arthritis Res. Ther. 2016, 18, 117. [Google Scholar] [CrossRef] [PubMed]
- Dominic, A.; Le, N.-T.; Takahashi, M. Loop between NLRP3 Inflammasome and Reactive Oxygen Species. Antioxidants Redox Signal. 2022, 36, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, S.M.; Grebe, K.; Whitehead, L.W.; Rogers, K.L.; Nebl, T.; Murphy, J.M.; Wicks, I.P. Monosodium Urate Crystals Generate Nuclease-Resistant Neutrophil Extracellular Traps via a Distinct Molecular Pathway. J. Immunol. 2018, 200, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Desai, J.; Steiger, S.; Anders, H.-J. Molecular Pathophysiology of Gout. Trends Mol. Med. 2017, 23, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Schauer, C.; Hoffmann, M.; Herrmann, M. Why does the gout attack stop? A roadmap for the immune pathogenesis of gout. RMD Open 2015, 1 (Suppl. S1), e000046. [Google Scholar] [CrossRef] [PubMed]
- Apel, F.; Zychlinsky, A.; Kenny, E.F. The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Cuevas, Y.; Hernández-Díaz, C.; Pineda, C.; Reginato, A.M.; Cerna-Cortés, J.F.; Ventura-Ríos, L.; López-Reyes, A. Molecular basis of oxidative stress in gouty arthropathy. Clin. Rheumatol. 2015, 34, 1667–1672. [Google Scholar] [CrossRef]
- Lepetsos, P.; Papavassiliou, K.A.; Papavassiliou, A.G. Redox and NF-κB signaling in osteoarthritis. Free. Radic. Biol. Med. 2018, 132, 90–100. [Google Scholar] [CrossRef]
- Trevisan, G.; Hoffmeister, C.; Rossato, M.F.; Oliveira, S.M.; Silva, M.A.; Silva, C.R.; Fusi, C.; Tonello, R.; Minocci, D.; Guerra, G.P.; et al. TRPA1 receptor stimulation by hydrogen peroxide is critical to trigger hyperalgesia and inflammation in a model of acute gout. Free. Radic. Biol. Med. 2014, 72, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. SnapShot: Reactive Oxygen Intermediates (ROI). Cell 2010, 140, 951. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, X.; Zhang, T.; Tian, K.; Li, Z.; Luo, F. Reactive oxygen species (ROS) scavenging biomaterials for anti-inflammatory diseases: From mechanism to therapy. J. Hematol. Oncol. 2023, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Manford, A.G.; Mena, E.L.; Shih, K.Y.; Gee, C.L.; McMinimy, R.; Martínez-González, B.; Sherriff, R.; Lew, B.; Zoltek, M.; Rodríguez-Pérez, F.; et al. Structural basis and regulation of the reductive stress response. Cell 2021, 184, 5375–5390. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Iv, C.D.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, R.; Yan, X.; Fan, K. Superoxide dismutase nanozymes: An emerging star for anti-oxidation. J. Mater. Chem. B 2021, 9, 6939–6957. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxid. Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef]
- Baker, A.; Lin, C.-C.; Lett, C.; Karpinska, B.; Wright, M.H.; Foyer, C.H. Catalase: A critical node in the regulation of cell fate. Free. Radic. Biol. Med. 2023, 199, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Davies, M.J.; Hawkins, C.L. Reactions and reactivity of myeloperoxidase-derived oxidants: Differential biological effects of hypochlorous and hypothiocyanous acids. Free. Radic. Res. 2012, 46, 975–995. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-W.; Zhang, Y.-N.; Wang, Y.-C.; Lu, Y.-B.; Huang, X.-L.; Lao, Y.-F.; Zhang, L.; Yang, J.; Shi, M.; Ma, H.-L. Myeloperoxidase: A new target for the treatment of stroke? Neural Regen. Res. 2022, 17, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Bartosz, G. The Cellular and Organismal Effects of Nitroxides and Nitroxide-Containing Nanoparticles. Int. J. Mol. Sci. 2024, 25, 1446. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I.; Lushchak, O.V. Interplay between reactive oxygen and nitrogen species in living organisms. Chem. Biol. Interact. 2021, 349, 109680. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef]
- De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; Bonora, E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. [Google Scholar] [CrossRef]
- Degechisa, S.T.; Dabi, Y.T.; Gizaw, S.T. The mitochondrial associated endoplasmic reticulum membranes: A platform for the pathogenesis of inflammation-mediated metabolic diseases. Immun. Inflamm. Dis. 2022, 10, e647. [Google Scholar] [CrossRef] [PubMed]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef]
- Madesh, M.; Hajnóczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Wang, L.; Hauenstein, A.V. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol. Asp. Med. 2020, 76, 100889. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Walle, L.V.; Lamkanfi, M. Drugging the NLRP3 inflammasome: From signalling mechanisms to therapeutic targets. Nat. Rev. Drug Discov. 2023, 23, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Akbal, A.; Dernst, A.; Lovotti, M.; Mangan, M.S.J.; McManus, R.M.; Latz, E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell. Mol. Immunol. 2022, 19, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2015, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxidants Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, P.; Feng, T.; Xie, Z.; Yang, M.; Chen, Z.; Hu, S.; Han, D.; Chen, W. Nrf2/HO-1 blocks TXNIP/NLRP3 interaction via elimination of ROS in oxygen-glucose deprivation-induced neuronal necroptosis. Brain Res. 2023, 1817, 148482. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, D.; Wu, L.; Zhang, J.; Wu, D.; Li, X.; Zhi, F.; Yang, G.; Kong, X.; Hong, J.; et al. Electroacupuncture inhibits the corneal ROS/TXNIP/NLRP3 signaling pathway in a rat model of dry eye syndrome. Acupunct. Med. 2022, 40, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- López-Reyes, A.; Medina-Luna, D.; Santamaría-Olmedo, M.; Martínez-Flores, K.; Zamudio-Cuevas, Y.; Fernández-Torres, J.; Martínez-Nava, G.A.; Olivos-Meza, A.; Camacho-Rea, C.; Fernández-Moreno, M.; et al. Soluble inflammatory mediators of synoviocytes stimulated by monosodium urate crystals induce the production of oxidative stress, pain, and inflammation mediators in chondrocytes: Secretome of synoviocytes induces chondrocyte damage. Clin. Rheumatol. 2021, 40, 3265–3271. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Zhou, X.; Qin, M.; Lin, Y.; Lin, J.; Chen, G.; Liu, A.; Ouyang, D.; Chen, D.; Pan, H. Corilagin Restrains NLRP3 Inflammasome Activation and Pyroptosis through the ROS/TXNIP/NLRP3 Pathway to Prevent Inflammation. Oxidative Med. Cell. Longev. 2022, 2022, 1652244. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, Z.; Bai, L.; Lei, S.; Zou, M.; Bao, Z.; Ren, Z.; Liu, K.; Gong, H.-H.; Ma, W.; et al. Piper longum L. ameliorates gout through the MAPK/PI3K-AKT pathway. J. Ethnopharmacol. 2024, 330, 118254. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, M.; Lu, Y.; Wang, M.; Xiao, J.; Xie, Q.; He, X.; Shuai, S. Sirt1 inhibits macrophage polarization and inflammation in gouty arthritis by inhibiting the MAPK/NF-κB/AP-1 pathway and activating the Nrf2/HO-1 pathway. Inflamm. Res. 2024, 73, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dai, A.; Yang, M.; Chen, S.; Deng, Z.; Li, L. p38MAPK Signaling Pathway in Osteoarthritis: Pathological and Therapeutic Aspects. J. Inflamm. Res. 2022, 15, 723–734. [Google Scholar] [CrossRef]
- Ronkina, N.; Gaestel, M. MAPK-Activated Protein Kinases: Servant or Partner? Annu. Rev. Biochem. 2022, 91, 505–540. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Wortzel, I.; Seger, R. Alternative Splicing of MAPKs in the Regulation of Signaling Specificity. Cells 2021, 10, 3466. [Google Scholar] [CrossRef]
- Jalmi, S.K.; Sinha, A.K. ROS mediated MAPK signaling in abiotic and biotic stress- striking similarities and differences. Front. Plant Sci. 2015, 6, 769. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Kim, S.; Chung, H.T.; Pae, H.O. Reactive oxygen species in the activation of MAP kinases. Method Enzym. 2013, 528, 27–48. [Google Scholar] [CrossRef]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. Reactive Oxygen Species-Induced Activation of the MAP Kinase Signaling Pathways. Antioxidants Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: From mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38MAPK: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell. Physiol. 2019, 234, 14951–14965. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Macleod, T.; Wong, C.; Harland, M.; McGonagle, D. Revisiting p38 Mitogen-Activated Protein Kinases (MAPK) in Inflammatory Arthritis: A Narrative of the Emergence of MAPK-Activated Protein Kinase Inhibitors (MK2i). Pharmaceuticals 2023, 16, 1286. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Lu, F.; Liu, L.; Yu, D.; Li, X.; Zhou, Q.; Liu, S. Therapeutic Effect of Rhizoma Dioscoreae Nipponicae on Gouty Arthritis Based on the SDF-1/CXCR 4 and p38 MAPK Pathway: An In Vivo and In Vitro Study. Phytotherapy Res. 2014, 28, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, S.; Liu, Y.; Kong, B.; Yan, W.; Jiang, T.; Tian, H.; Liu, Z.; Shi, Q.; Wang, Y.; et al. Cynarin suppresses gouty arthritis induced by monosodium urate crystals. Bioengineered 2022, 13, 11782–11793. [Google Scholar] [CrossRef]
- Panipinto, P.M.; Singh, A.K.; Shaikh, F.S.; Siegel, R.J.; Chourasia, M.; Ahmed, S. Takinib Inhibits Inflammation in Human Rheumatoid Arthritis Synovial Fibroblasts by Targeting the Janus Kinase-Signal Transducer and Activator of Transcription 3 (JAK/STAT3) Pathway. Int. J. Mol. Sci. 2021, 22, 12580. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Li, Z.; Zhang, S.; Zhang, J.; Jia, E. Novel perception of neutrophil extracellular traps in gouty inflammation. Int. Immunopharmacol. 2023, 115, 109642. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef]
- De Mattos, T.R.F.; Formiga-Jr, M.A.; Saraiva, E.M. Resveratrol prevents the release of neutrophil extracellular traps (NETs) by controlling hydrogen peroxide levels and nuclear elastase migration. Sci. Rep. 2024, 14, 9107. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Yu, P.; Chu, C.; Wang, Z.; Xu, W.; Cheng, F.; Zhao, H.; Qiu, Z. Magnesium hydride attenuates intestinal barrier injury during hemorrhage shock by regulating neutrophil extracellular trap formation via the ROS/MAPK/PAD4 pathway. Int. Immunopharmacol. 2024, 130, 111688. [Google Scholar] [CrossRef]
- Maueröder, C.; Kienhöfer, D.; Hahn, J.; Schauer, C.; Manger, B.; Schett, G.; Herrmann, M.; Hoffmann, M.H. How neutrophil extracellular traps orchestrate the local immune response in gout. J. Mol. Med. 2015, 93, 727–734. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Xiang, M.; Yin, M.; Xie, S.; Shi, L.; Nie, W.; Shi, B.; Yu, G. The molecular mechanism of neutrophil extracellular traps and its role in bone and joint disease. Heliyon 2023, 9, e22920. [Google Scholar] [CrossRef]
- Azzouz, D.; Khan, M.A.; Palaniyar, N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. 2021, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef] [PubMed]
- Jia, E.; Li, Z.; Geng, H.; Zhu, H.; Wang, Y.; Lin, F.; Jiang, Y.; Zhang, J. Neutrophil extracellular traps induce the bone erosion of gout. BMC Musculoskelet. Disord. 2022, 23, 1128. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Yang, D.; Feng, F.; Zhu, D.; Li, X. miR-3146 induces neutrophil extracellular traps to aggravate gout flare. J. Clin. Lab. Anal. 2021, 35, e24032. [Google Scholar] [CrossRef] [PubMed]
- Fousert, E.; Toes, R.; Desai, J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells 2020, 9, 915. [Google Scholar] [CrossRef]
- Euler, M.; Hoffmann, M.H. The double-edged role of neutrophil extracellular traps in inflammation. Biochem. Soc. Trans. 2019, 47, 1921–1930. [Google Scholar] [CrossRef]
- Tao, H.; Mo, Y.; Liu, W.; Wang, H. A review on gout: Looking back and looking ahead. Int. Immunopharmacol. 2023, 117, 109977. [Google Scholar] [CrossRef]
- Liu, L.; Shan, L.; Wang, H.; Schauer, C.; Schoen, J.; Zhu, L.; Lu, C.; Wang, Z.; Xue, Y.; Wu, H.; et al. Neutrophil Extracellular Trap–Borne Elastase Prevents Inflammatory Relapse in Intercritical Gout. Arthritis Rheumatol. 2023, 75, 1039–1047. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Cleophas, M.C.; Crişan, T.O.; Joosten, L.A. Factors modulating the inflammatory response in acute gouty arthritis. Curr. Opin. Rheumatol. 2017, 29, 163–170. [Google Scholar] [CrossRef]
- Fan, W.; Chen, S.; Wu, X.; Zhu, J.; Li, J. Resveratrol Relieves Gouty Arthritis by Promoting Mitophagy to Inhibit Activation of NLRP3 Inflammasomes. J. Inflamm. Res. 2021, 14, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Chen, W.-Y.; Yu, C.-L.; Tsai, C.-Y.; Hsieh, S.-C. Gouty arthritis involves impairment of autophagic degradation via cathepsin D inactivation-mediated lysosomal dysfunction that promotes apoptosis in macrophages. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166703. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z. ROS-induced lipid peroxidation modulates cell death outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Bisaglia, M.; Plotegher, N. Linking ROS Levels to Autophagy: The Key Role of AMPK. Antioxidants 2023, 12, 1406. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhang, Y.; Wang, Z.; Wang, S.; Jiang, X.; Cui, H.; Zhou, T.; He, Z.; Feng, H.; Guo, Q.; et al. ATM at the crossroads of reactive oxygen species and autophagy. Int. J. Biol. Sci. 2021, 17, 3080–3090. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, X.-Y.; Liu, Y.-J.; Feng, J.; Wu, Y.; Shen, H.-M.; Lu, G.-D. Full-coverage regulations of autophagy by ROS: From induction to maturation. Autophagy 2021, 18, 1240–1255. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Zhang, Q.B.; Zheng, J.X.; Jian, G.L.; Liu, T.H.; He, X.; Xiao, F.N.; Xiong, Q.; Qing, Y.F. POS0136 roles of autophagy in the pathogenesis of primary gouty arthritis. Ann. Rheum. Dis. 2021, 80 (Suppl. S1), 280. [Google Scholar] [CrossRef]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef]
- Navaneethan, R.D.; Ncj, P.L.; Ramaiah, M.; Ravindran, R.; Chinnathambi, A.; Alharbi, S.A.; Sivagnanam, A.; Mohemedibrahim, P. Caralluma pauciflora based Ag-NPs activate ROS-induced apoptosis through down-regulation of AKT, mTOR and pI3K signaling in human Gastric Cancer (AGS) cells. Nanotechnology 2024, 35, 195102. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-F.; Su, G.; Chen, L.-X.; Zhou, J.-P.; Gao, J.; Zhang, J.-J.; Wu, Q.-H.; Chen, W.; Chen, D.-Y.; Zhang, Z.-C. Irisin Attenuates Apoptosis Following Ischemia–Reperfusion Injury Through Improved Mitochondria Dynamics and ROS Suppression Mediated Through the PI3K/Akt/mTOR Axis. Mol. Neurobiol. 2023, 60, 4261–4272. [Google Scholar] [CrossRef]
- Hsieh, C.-Y.; Li, L.-H.; Lam, Y.; Fang, Z.; Gan, C.H.; Rao, Y.K.; Chiu, H.-W.; Wong, W.-T.; Ju, T.-C.; Chen, F.-H.; et al. Synthetic 4-Hydroxy Auxarconjugatin B, a Novel Autophagy Inducer, Attenuates Gouty Inflammation by Inhibiting the NLRP3 Inflammasome. Cells 2020, 9, 279. [Google Scholar] [CrossRef]
- Chang, S.; Tang, M.; Zhang, B.; Xiang, D.; Li, F. Ferroptosis in inflammatory arthritis: A promising future. Front. Immunol. 2022, 13, 955069. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2020, 17, 2054–2081. [Google Scholar] [CrossRef]
- Dixon, S.J.; Olzmann, J.A. The cell biology of ferroptosis. Nat. Rev. Mol. Cell Biol. 2024, 25, 424–442. [Google Scholar] [CrossRef]
- Dixon, S.J.; Pratt, D.A. Ferroptosis: A flexible constellation of related biochemical mechanisms. Mol. Cell 2023, 83, 1030–1042. [Google Scholar] [CrossRef]
- Li, X.; He, T.; Yu, K.; Lu, Q.; Alkasir, R.; Guo, G.; Xue, Y. Markers of Iron Status Are Associated with Risk of Hyperuricemia among Chinese Adults: Nationwide Population-Based Study. Nutrients 2018, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- DeVallance, E.R.; Schmidt, H.M.; Seman, M.; Lewis, S.E.; Wood, K.C.; Vickers, S.D.; Hahn, S.A.; Velayutham, M.; Hileman, E.A.; Vitturi, D.A.; et al. Hemin and iron increase synthesis and trigger export of xanthine oxidoreductase from hepatocytes to the circulation. Redox Biol. 2023, 67, 102866. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.-C.; Sun, W.-K.; Deng, Q.-Q.; Cheng, L.; Huang, X.; Hu, L.-K.; Li, H.-N. Identification of Key lncRNAs in Gout Under Copper Death and Iron Death Mechanisms: A Study Based on ceRNA Network Analysis and Random Forest Algorithm. Mol. Biotechnol. 2024, 66. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, F.; Zhong, S.; Zhao, K.; Liao, H.; Liang, J.; Zheng, Q.; Wu, H.; Zhang, S.; Cao, Y.; et al. Protecting against ferroptosis in hyperuricemic nephropathy: The potential of ferrostatin-1 and its inhibitory effect on URAT1. Eur. J. Pharmacol. 2024, 971, 176528. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Murakami, S.; Kusano, Y.; Okazaki, K.; Akaike, T.; Motohashi, H. NRF2 signalling in cytoprotection and metabolism. Br. J. Pharmacol. 2023, 181, 16246. [Google Scholar] [CrossRef]
- Badawy, A.M.; Taha, M.; Elazab, S.T.; El-Shenbaby, I.; Alghamdi, B.A.; Al-Kushi, A.G.; Fathy, K.; Baokbah, T.A.S.; Ibrahim, M.M. Targeting of Nrf2/PPARγ/NLRP3 Signaling Pathway by Stevia rebudiana Bertoni Extract Provides a Novel Insight into Its Protective Effect against Acute Gouty Arthritis-Induced Synovial Inflammation, Oxidative Stress and Apoptosis in a Rat Model. Processes 2022, 10, 1751. [Google Scholar] [CrossRef]
- Catalán, L.; Carceller, M.C.; Terencio, M.C.; Alcaraz, M.J.; Ferrándiz, M.L.; Montesinos, M.C. Osteostatin Mitigates Gouty Arthritis through the Inhibition of Caspase-1 Activation and Upregulation of Nrf2 Expression. Int. J. Mol. Sci. 2024, 25, 2752. [Google Scholar] [CrossRef] [PubMed]
- Suntres, Z.E.; Coccimiglio, J.; Alipour, M. The Bioactivity and Toxicological Actions of Carvacrol. Crit. Rev. Food Sci. Nutr. 2015, 55, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Al Kury, L.T.; Atzaz, N.; Alattar, A.; Alshaman, R.; Shah, F.A.; Li, S. Carvacrol Alleviates Hyperuricemia-Induced Oxidative Stress and Inflammation by Modulating the NLRP3/NF-κB Pathwayt. Drug Des. Dev. Ther. 2022, 16, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Akash, S.R.; Tabassum, A.; Aditee, L.M.; Rahman, A.; Hossain, I.; Hannan, A.; Uddin, J. Pharmacological insight of rutin as a potential candidate against peptic ulcer. Biomed. Pharmacother. 2024, 177, 116961. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Huang, J.; Li, Y.; Lin, Z.; Zhang, B. Rutin ameliorates gout via reducing XOD activity, inhibiting ROS production and NLRP3 inflammasome activation in quail. Biomed. Pharmacother. 2023, 158, 114175. [Google Scholar] [CrossRef]
- Zhao, F.-Y.; Spencer, S.J.; Kennedy, G.A.; Zheng, Z.; Conduit, R.; Zhang, W.-J.; Xu, P.; Yue, L.-P.; Wang, Y.-M.; Xu, Y.; et al. Acupuncture for primary insomnia: Effectiveness, safety, mechanisms and recommendations for clinical practice. Sleep Med. Rev. 2024, 74, 101892. [Google Scholar] [CrossRef] [PubMed]
- Ulett, G.A.; Han, S.; Han, J.-S. Electroacupuncture: Mechanisms and clinical application. Biol. Psychiatry 1998, 44, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, B.; Yin, C.; Zeng, D.; Nie, H.; Li, Y.; Tai, Y.; He, X.; Liu, B. Electroacupuncture improves gout arthritis pain via attenuating ROS-mediated NLRP3 inflammasome overactivation. Chin. Med. 2023, 18, 86. [Google Scholar] [CrossRef]
- Ichihara, H.; Hino, M.; Makizono, T.; Umebayashi, M.; Matsumoto, Y.; Ueoka, R. Inhibitory effects of hybrid liposomes on the growth of synoviocyte causing rheumatoid arthritis. Bioorganic Med. Chem. Lett. 2011, 21, 207–210. [Google Scholar] [CrossRef]
- Li, C.; Wang, C.; Guo, Y.; Wen, R.; Yan, L.; Zhang, F.; Gong, Q.; Yu, H. Research on the effect and underlying molecular mechanism of Cangzhu in the treatment of gouty arthritis. Eur. J. Pharmacol. 2022, 927, 175044. [Google Scholar] [CrossRef]
- Yan, Y.; Yu, L.; Chen, B.; Cao, C.; Zhao, H.; Wang, Q.; Xie, D.; Xi, Y.; Zhang, C.; Cheng, J. Mastoparan M Suppressed NLRP3 Inflammasome Activation by Inhibiting MAPK/NF-κB and Oxidative Stress in Gouty Arthritis. J. Inflamm. Res. 2023, 16, 6179–6193. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, C.; Li, F.; Xu, W.; Zhang, X.; Huang, Y.; Xia, D. Targeting Neutrophil Extracellular Traps in Gouty Arthritis: Insights into Pathogenesis and Therapeutic Potential. J. Inflamm. Res. 2024, 17, 1735–1763. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, J.; Guo, Y.; Li, M.; Yang, K.; Liu, Y.; Ge, D.; Liu, Y.; Xue, C.; Xia, T.; et al. Monosodium Urate Crystal-Induced Pyroptotic Cell Death in Neutrophil and Macrophage Facilitates the Pathological Progress of Gout. Small 2023, 20, e2308749. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, L.; Sun, H.; Liu, S. Relief of gouty arthritis in rats by total saponins from Dioscorea nipponica Makino through suppression of neutrophil extracellular trap formation via the PI3K/AKT/mTOR axis. Exp. Ther. Med. 2023, 26, 447. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsuchiya, M.; Yoshida, S.; Ogawa, K.; Chen, W.; Kanzaki, M.; Takahashi, T.; Fujita, R.; Li, Y.; Yabe, Y.; et al. Tissue accumulation of neutrophil extracellular traps mediates muscle hyperalgesia in a mouse model. Sci. Rep. 2022, 12, 4136. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, E.; Gamberucci, A.; Lucherini, O.-M.; Alì, A.; Simpatico, A.; Lorenzini, S.; Lazzerini, P.-E.; Tripodi, S.; Frediani, B.; Selvi, E. Neutrophil extracellular traps release in gout and pseudogout depends on the number of crystals regardless of leukocyte count. Rheumatology 2021, 60, 4920–4928. [Google Scholar] [CrossRef]
- Mandell, B.F.; Yeo, A.E.; Lipsky, P.E. Tophus resolution in patients with chronic refractory gout who have persistent urate-lowering responses to pegloticase. Arthritis Res. Ther. 2018, 20, 286. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.; Zhang, X.; Jiang, C.; Zhang, F.; Xu, C.; Fang, S.; Shang, X.; Zhang, J.; Yin, Z. 3β,23-Dihydroxy-12-ene-28-ursolic Acid Isolated from Cyclocarya paliurus Alleviates NLRP3 Inflammasome-Mediated Gout via PI3K-AKT-mTOR-Dependent Autophagy. Evidence-Based Complement. Altern. Med. 2022, 2022, 5541232. [Google Scholar] [CrossRef]
- Han, J.; Shi, G.; Li, W.; Wang, S.; Bai, J.; Sun, X.; Xie, Y.; Sui, F.; Chen, F.; Jiang, D. Zisheng Shenqi Decoction Ameliorates Monosodium Urate-Mediated Gouty Arthritis in Rats via Promotion of Autophagy through the AMPK/mTOR Signaling Pathway. Evid.-Based Complement. Altern. Med. 2021, 2021, 6918026. [Google Scholar] [CrossRef]
- Subawa, I.; Astawa, P.; Bakta, I.; Astawa, I.M.; Krisna, G.A. Purple sweet potato (Ipomoea batatas L.) extract effects on levels of inflammatory markers and chondrocyte count in gout arthritis Wistar rat model. Foot Ankle Surg. 2023, 29, 611–615. [Google Scholar] [CrossRef]
- Chen, Q.M. Nrf2 for cardiac protection: Pharmacological options against oxidative stress. Trends Pharmacol. Sci. 2021, 42, 729–744. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, D.; Yin, C.; Wei, H.; Li, Y.; Yang, Y.; Nie, H.; Pan, Y.; Xu, R.; Tai, Y.; Du, J.; et al. Activation of Nrf2 antioxidant signaling alleviates gout arthritis pain and inflammation. Biomed. Pharmacother. 2024, 170, 115957. [Google Scholar] [CrossRef]
- Shi, K.; Zhou, S.; Lei, L.; Huang, L.; Zhang, C.; Shao, H. Chemical Composition and Phytotoxic Activity of Artemisia selengensis Turcz. Volatiles. Chem. Biodivers. 2021, 18, e2100701. [Google Scholar] [CrossRef]
- Cao, W.; Wu, T.; Liang, F.; Fang, Y.; Cheng, Y.; Pan, S.; Xu, X. Protective effects of di-caffeoylquinic acids from Artemisia selengensis Turcz leaves against monosodium urate-induced inflammation via the modulation of NLRP3 inflammasome and Nrf2 signaling pathway in THP-1 macrophages. J. Food Biochem. 2022, 46, e14252. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Zhong, X.; Xia, H.; Zhao, M.; Zhao, M.; Xu, L.; Guo, X.; You, C.-G. Lipoxin A4 attenuates MSU-crystal-induced NLRP3 inflammasome activation through suppressing Nrf2 thereby increasing TXNRD2. Front. Immunol. 2022, 13, 1060441. [Google Scholar] [CrossRef]
- Ruiz-Miyazawa, K.W.; Pinho-Ribeiro, F.A.; Borghi, S.M.; Staurengo-Ferrari, L.; Fattori, V.; Amaral, F.A.; Teixeira, M.M.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; et al. Hesperidin Methylchalcone Suppresses Experimental Gout Arthritis in Mice by Inhibiting NF-κB Activation. J. Agric. Food Chem. 2018, 66, 6269–6280. [Google Scholar] [CrossRef]
- Qin, D.-E.; Liang, W.; Yu, Y.; Whelan, E.C.; Yuan, X.; Wang, Z.-L.; Wu, X.-W.; Cao, Z.-R.; Hua, S.-Y.; Yin, L.; et al. Modified Simiaowan prevents and treats gouty arthritis via the Nrf2/NLRP3 inflammasome signaling pathway. J. Ethnopharmacol. 2024, 318, 116906. [Google Scholar] [CrossRef]
- Cheng, J.-J.; Ma, X.-D.; Ai, G.-X.; Yu, Q.-X.; Chen, X.-Y.; Yan, F.; Li, Y.-C.; Xie, J.-H.; Su, Z.-R.; Xie, Q.-F. Palmatine Protects Against MSU-Induced Gouty Arthritis via Regulating the NF-κB/NLRP3 and Nrf2 Pathways. Drug Des. Dev. Ther. 2022, 16, 2119–2132. [Google Scholar] [CrossRef]
- Lin, Y.; Luo, T.; Weng, A.; Huang, X.; Yao, Y.; Fu, Z.; Li, Y.; Liu, A.; Li, X.; Chen, D.; et al. Gallic Acid Alleviates Gouty Arthritis by Inhibiting NLRP3 Inflammasome Activation and Pyroptosis Through Enhancing Nrf2 Signaling. Front. Immunol. 2020, 11, 580593. [Google Scholar] [CrossRef]
- Dinesh, P.; Rasool, M. Berberine, an isoquinoline alkaloid suppresses TXNIP mediated NLRP3 inflammasome activation in MSU crystal stimulated RAW 264.7 macrophages through the upregulation of Nrf2 transcription factor and alleviates MSU crystal induced inflammation in rats. Int. Immunopharmacol. 2017, 44, 26–37. [Google Scholar] [CrossRef]
- Qadri, M.; Khired, Z.; Alaqi, R.; Elsayed, S.; Alarifi, A.; Ahmed, R.; Alhamami, H.; Khardali, A.; Hakami, W. Zerumbone reduces TLR2 stimulation-induced M1 macrophage polarization pattern via upregulation of Nrf-2 expression in murine macrophages. Saudi Pharm. J. 2024, 32, 101956. [Google Scholar] [CrossRef]
- Zhou, X.; Qin, M.; He, L.; Zhang, Y.; Liu, A.; Chen, D.; Pan, H. Geraniin restricts inflammasome activation and macrophage pyroptosis by preventing the interaction between ASC and NLRP3 to exert anti-inflammatory effects. Int. Immunopharmacol. 2024, 129, 111656. [Google Scholar] [CrossRef] [PubMed]
- Charoenwutthikun, S.; Chanjitwiriya, K.; Roytrakul, S.; Kunthalert, D. A wild rice-derived peptide R14 ameliorates monosodium urate crystals-induced IL-1β secretion through inhibition of NF-κB signaling and NLRP3 inflammasome activation. PeerJ 2023, 11, e15295. [Google Scholar] [CrossRef]
- Cao, Y.; Hu, Y.; Jin, X.-F.; Liu, Y.; Zou, J.-M. Dimethyl fumarate attenuates MSU-induced gouty arthritis by inhibiting NLRP3 inflammasome activation and oxidative stress. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 628–641. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, H.; Li, X. The protective effects of S14G-humanin (HNG) against mono-sodium urate (MSU) crystals- induced gouty arthritis. Bioengineered 2021, 13, 345–356. [Google Scholar] [CrossRef]
- Zhou, X.; Shi, Q.; Li, J.; Quan, S.; Zhang, X.; Gu, L.; Li, H.; Ju, Y.; Hu, M.; Li, Q. Medicinal fungus Phellinus igniarius alleviates gout in vitro by modulating TLR4/NF-kB/NLRP3 signaling. Front. Pharmacol. 2022, 13, 1011406. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Huang, X.; Tang, Y.; Li, J.; Tan, Y.; Yuan, Q. Effects of evodiamine on ROS/TXNIP/NLRP3 pathway against gouty arthritis. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 397, 1015–1023. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Y.; Wang, T.; Sun, Y.; Cai, S. The preventive effect of Chinese sumac fruit against monosodium urate-induced gouty arthritis in rats by regulating several inflammatory pathways. Food Funct. 2023, 14, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Jati, G.A.K.; Assihhah, N.; Wati, A.A.; Salasia, S.I.O. Immunosuppression by piperine as a regulator of the NLRP3 inflammasome through MAPK/NF-κB in monosodium urate-induced rat gouty arthritis. Veter- World 2022, 15, 288–298. [Google Scholar] [CrossRef]
- Liu, H.-J.; Pan, X.-X.; Liu, B.-Q.; Gui, X.; Hu, L.; Jiang, C.-Y.; Han, Y.; Fan, Y.-X.; Tang, Y.-L.; Liu, W.-T. Grape seed-derived procyanidins alleviate gout pain via NLRP3 inflammasome suppression. J. Neuroinflammat. 2017, 14, 74. [Google Scholar] [CrossRef]
- Li, C.; Huang, Y.; Wu, C.; Qiu, Y.; Zhang, L.; Xu, J.; Zheng, J.; Zhang, X.; Li, F.; Xia, D. Astilbin inhibited neutrophil extracellular traps in gouty arthritis through suppression of purinergic P2Y6 receptor. Phytomedicine 2024, 130, 155754. [Google Scholar] [CrossRef] [PubMed]
- McWherter, C.; Choi, Y.-J.; Serrano, R.L.; Mahata, S.K.; Terkeltaub, R.; Liu-Bryan, R. Arhalofenate acid inhibits monosodium urate crystal-induced inflammatory responses through activation of AMP-activated protein kinase (AMPK) signaling. Arthritis Res. Ther. 2018, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Yang, Z.; Ou, Y.; Liang, S.; Deng, W.; Chen, H.; Zhang, C.; Hua, L.; Hu, W.; Sun, P. Tanshinones inhibit NLRP3 inflammasome activation by alleviating mitochondrial damage to protect against septic and gouty inflammation. Int. Immunopharmacol. 2021, 97, 107819. [Google Scholar] [CrossRef]
- Jiang, H.; Song, D.; Zhou, X.; Chen, F.; Yu, Q.; Ren, L.; Dai, Q.; Zeng, M. Maresin1 ameliorates MSU crystal-induced inflammation by upregulating Prdx5 expression. Mol. Med. 2023, 29, 158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Sun, D.; He, Y.; Jiang, Y.; Cheng, K.-W.; Chen, F. DHA protects against monosodium urate-induced inflammation through modulation of oxidative stress. Food Funct. 2019, 10, 4010–4021. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, D.M.; Kabil, S.L. Ozone Therapy Alleviates Monosodium Urate Induced Acute Gouty Arthritis in Rats Through Inhibition of NLRP3 Inflammasome. Curr. Drug Ther. 2021, 16, 345–353. [Google Scholar] [CrossRef]
- Yin, C.; Liu, B.; Wang, P.; Li, X.; Li, Y.; Zheng, X.; Tai, Y.; Wang, C.; Liu, B. Eucalyptol alleviates inflammation and pain responses in a mouse model of gout arthritis. Br. J. Pharmacol. 2020, 177, 2042–2057. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, M.; Lugrin, J.; Ehirchiou, D.; Nasi, S.; Ishii, I.; So, A.; Martinon, F.; Busso, N. Hydrogen sulfide inhibits NLRP3 inflammasome activation and reduces cytokine production both in vitro and in a mouse model of inflammation. J. Biol. Chem. 2018, 293, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Zhou, Q.; Wang, Z.; Mo, C.; Dong, W.; Wei, N.; Zhong, W.; You, Y.; Wang, Y.; Wang, Z. Polydatin ameliorates inflammation and oxidative stress associated with MSU-induced gouty arthritis in mice by regulating PPAR-γ and ferritin activation. Life Sci. 2023, 326, 121766. [Google Scholar] [CrossRef]
- Bardin, T.; Richette, P. Impact of comorbidities on gout and hyperuricaemia: An update on prevalence and treatment options. BMC Med. 2017, 15, 123. [Google Scholar] [CrossRef]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; Brignardello-Petersen, R.; Guyatt, G.; Abeles, A.M.; Gelber, A.C.; Harrold, L.R.; Khanna, D.; King, C.; et al. 2020 American College of Rheumatology Guideline for the Management of Gout. Arthritis Rheumatol. 2020, 72, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Mikuls, T.R. Gout. New Engl. J. Med. 2022, 387, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Qaseem, A.; Harris, R.P.; Forciea, M.A.; Denberg, T.D.; Barry, M.J.; Boyd, C.; Chow, R.D.; Humphrey, L.L.; Kansagara, D.; Vijan, S.; et al. Management of Acute and Recurrent Gout: A Clinical Practice Guideline From the American College of Physicians. Ann. Intern. Med. 2017, 166, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Shekelle, P.G.; Newberry, S.J.; FitzGerald, J.D.; Motala, A.; O’Hanlon, C.E.; Tariq, A.; Okunogbe, A.; Han, D.; Shanman, R. Management of Gout: A Systematic Review in Support of an American College of Physicians Clinical Practice Guideline. Ann. Intern. Med. 2017, 166, 37–51. [Google Scholar] [CrossRef]
- Van Durme, C.M.; Wechalekar, M.D.; Landewé, R.B.; Pardo, J.P.; Cyril, S.; van der Heijde, D.; Buchbinder, R. Non-steroidal anti-inflammatory drugs for acute gout. Cochrane Database Syst. Rev. 2021, 2021, CD010120. [Google Scholar] [CrossRef]
- McKenzie, B.J.; Wechalekar, M.D.; Johnston, R.V.; Schlesinger, N.; Buchbinder, R. Colchicine for acute gout. Cochrane Database Syst. Rev. 2021, 2021, CD006190. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Y.; Huang, Q.; Deng, W.; Li, T.W. POS0133 monosodium urate crystals reduce human ligament cells viability through increase of ros production. Ann. Rheum. Dis. 2021, 80 (Suppl. S1), 278. [Google Scholar] [CrossRef]
- Halliwell, B. Understanding mechanisms of antioxidant action in health and disease. Nat. Rev. Mol. Cell Biol. 2023, 25, 13–33. [Google Scholar] [CrossRef]
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2023, 23, 274–288. [Google Scholar] [CrossRef]
- Spel, L.; Martinon, F. Inflammasomes contributing to inflammation in arthritis. Immunol. Rev. 2020, 294, 48–62. [Google Scholar] [CrossRef]
- Xiao, L.; Lin, S.; Xu, W.; Sun, E. Downregulation of Sox8 mediates monosodium urate crystal-induced autophagic impairment of cartilage in gout arthritis. Cell Death Discov. 2023, 9, 95. [Google Scholar] [CrossRef]
- Dalbeth, N.; Haskard, D.O. Mechanisms of inflammation in gout. Rheumatology 2005, 44, 1090–1096. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Zhu, G.; Zhang, Y.; He, M.; Zhang, J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp. Cell Res. 2016, 341, 42–53. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Tan, J.; Ye, J.; Cui, W.; Hou, J.; Liu, P.; Li, J.; Wang, S.; Zhao, Q. Anti-inflammation is an important way that Qingre-Huazhuo-Jiangsuan recipe treats acute gouty arthritis. Front. Pharmacol. 2023, 14, 1268641. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.; Lin, S.; Guan, W.; Liang, H.; Shen, J. Neutrophil autophagy induced by monosodium urate crystals facilitates neutrophil extracellular traps formation and inflammation remission in gouty arthritis. Front. Endocrinol. 2023, 14, 1071630. [Google Scholar] [CrossRef]
- Skendros, P.; Mitroulis, I.; Ritis, K. Autophagy in Neutrophils: From Granulopoiesis to Neutrophil Extracellular Traps. Front. Cell Dev. Biol. 2018, 6, 109. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil Extracellular Trap Formation Is Associated with IL-1β and Autophagy-Related Signaling in Gout. PLoS ONE 2011, 6, e29318. [Google Scholar] [CrossRef]
- Yin, C.; Liu, B.; Li, Y.; Li, X.; Wang, J.; Chen, R.; Tai, Y.; Shou, Q.; Wang, P.; Shao, X.; et al. IL-33/ST2 induces neutrophil-dependent reactive oxygen species production and mediates gout pain. Theranostics 2020, 10, 12189–12203. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Target | Mechanisms | Therapeutic Effect | Bibliography |
|---|---|---|---|
| ROS-NLRP3 | ROS activate NLRP3 inflammatory vesicles, release inflammatory factors, and in turn induce ROS production | Reduces ROS production, attenuates the association between ROS and NLRP3, disrupts the vicious cycle of oxidation and inflammation, and relieves inflammation | [175,176,177,178,179,180,181,182,183,184,185,186,187] |
| ROS-MAPK | ROS activate multiple pathways in the MAPK cascade to further activate MAPK, ultimately inducing inflammation and apoptosis | Reduces ROS production and disrupts the upstream activation pathway of MAPK to alleviate cellular inflammation and apoptosis | [160,188,189,190] |
| ROS-NET | ROS induce NETosis, promote cell rupture, release DAMPs, and trigger inflammatory effects. Massive release of reticulocyte contents, as the material basis of tophus | Reduces ROS production, prevents NETosis from upstream mechanisms, relieves inflammatory response, and prevents tophus formation | [191] |
| ROS-autophagy | ROS activate autophagy upstream via multiple pathways, and autophagy further reduces ROS and inflammation by phagocytosis of damaged mitochondria and other organelles | Reduces ROS production and promotes autophagosome formation to further alleviate oxidative stress and inflammation formation | [134,192,193] |
| ROS-Ferroptosis | Intracellular free iron or iron-containing enzymes react with the lipids of PUFA and ROS to produce high levels of lipid peroxides, thereby disrupting the biophysical properties of the plasma membrane leading to rupture of the cell membrane and cell death | Inhibits the production of ROS, reduces the formation of lipid peroxides, and prevents the rupture of cell membranes due to peroxidation, leading to cell death | No treatment has been reported. |
| ROS-Nrf2 | ROS activation upstream signaling promotes Nrf2 nuclear translocation, the formation of multiple antioxidant proteins, and the subsequent degradation of excess intracellular ROS | Inhibition of ROS production, activation of Nrf2, promotion of nuclear translocation, and further degradation of ROS | [194,195,196,197,198,199] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Li, D.; Fan, M.; Yuan, J.; Xie, C.; Yuan, H.; Xie, H.; Gao, H. Mechanism of Reactive Oxygen Species-Guided Immune Responses in Gouty Arthritis and Potential Therapeutic Targets. Biomolecules 2024, 14, 978. https://doi.org/10.3390/biom14080978
Zhang S, Li D, Fan M, Yuan J, Xie C, Yuan H, Xie H, Gao H. Mechanism of Reactive Oxygen Species-Guided Immune Responses in Gouty Arthritis and Potential Therapeutic Targets. Biomolecules. 2024; 14(8):978. https://doi.org/10.3390/biom14080978
Chicago/Turabian StyleZhang, Sai, Daocheng Li, Mingyuan Fan, Jiushu Yuan, Chunguang Xie, Haipo Yuan, Hongyan Xie, and Hong Gao. 2024. "Mechanism of Reactive Oxygen Species-Guided Immune Responses in Gouty Arthritis and Potential Therapeutic Targets" Biomolecules 14, no. 8: 978. https://doi.org/10.3390/biom14080978