
Synthesis and Biological Profiling of Seven Heparin and Heparan Sulphate Analogue Trisaccharides

 ,
,  , ,
, ,  , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
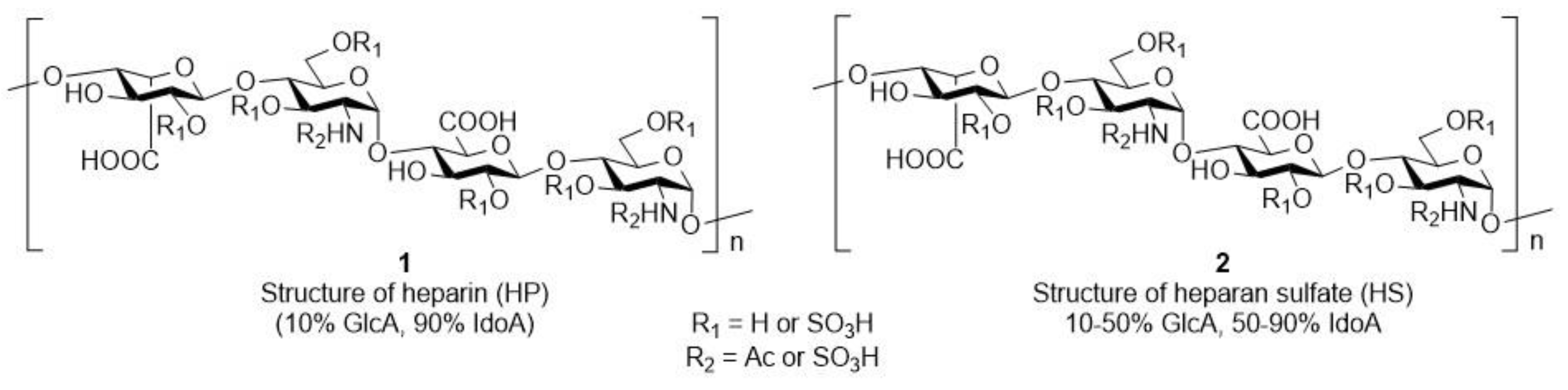
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. General Methods
General Method A for Zemplén Deacetylation (10 and 12)
General Method B for Cleavage of the Benzyl Groups by Catalytic Hydrogenation (11 and 33)
General Method C for Introduction of the Sulphate Ester Groups (13 and 15)
General Method D for Deacetylation/Debenzoylation of the Sulphated Derivatives (14 and 15)
General Method E for Glycosylation Reactions Using Thioglycoside Donor (18, 25 and 30)
General Method F for Regioselective Ring Opening Reaction of the 4,6-O-acetals (19 and 26)
General Method G for Cleavage of the (2-Naphthyl)methyl Group (27 and 31)
General Method H for Oxidation of the C-6-OH to Carboxylic Acid (28 and 32)
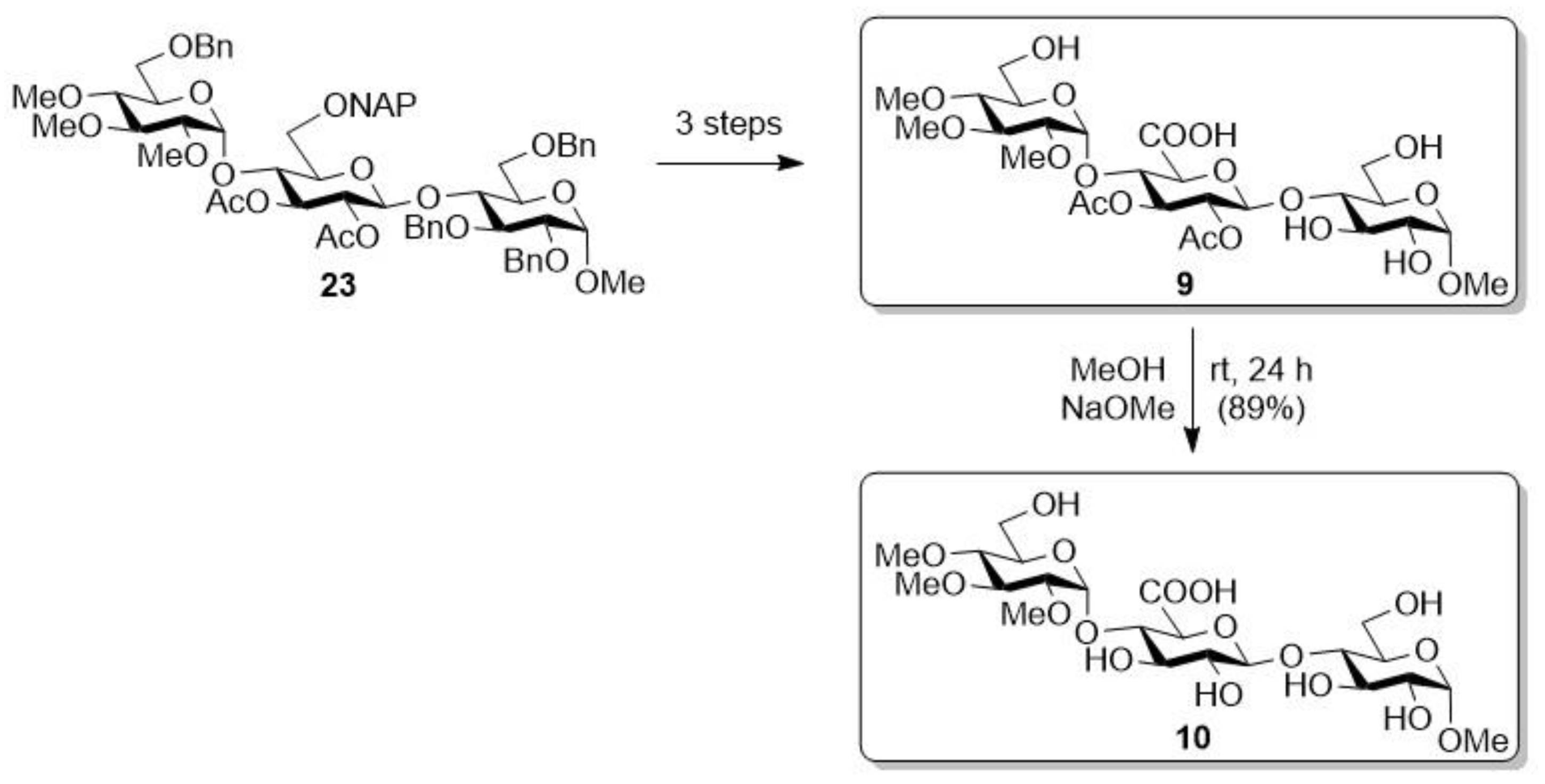
2.1.2. Sodium [methyl (2,3,4-tri-O-methyl-α-d-glucopyranosyl)-(1→4)-(β-d-glucopyranosyl-uronate)-(1→4)-α-d-glucopyranoside] (10)
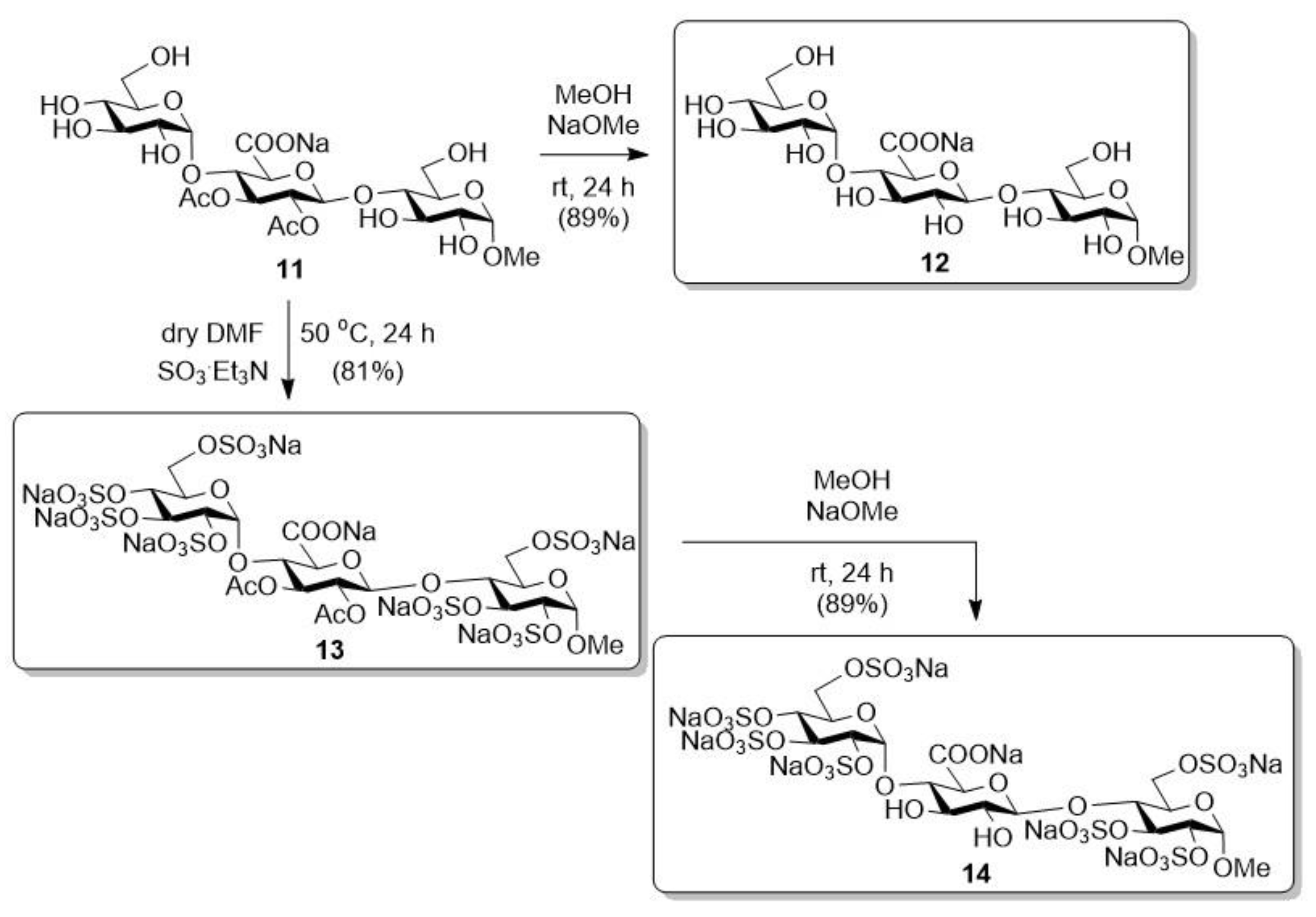
2.1.3. Sodium [methyl (α-d-glucopyranosyl)-(1→4)-(2,3-di-O-acetyl-β-d-glucopyranosyl-uronate)-(1→4)-α-d-glucopyranoside] (11)
2.1.4. Sodium [methyl (α-d-glucopyranosyl)-(1→4)-(β-d-glucopyranosyl-uronate)-(1→4)-α-d-glucopyranoside] (12)
2.1.5. Octa Sodium [methyl (2,3,4,6-tetra-O-sulfonato-α-d-glucopyranosyl)-(1→4)-(2,3-di-O-acetyl-β-d-glucopyranosyl-uronate)-(1→4)-2,3,6-tri-O-sulfonato-α-d-glucopyranoside] (13)
2.1.6. Octa Sodium [methyl (2,3,4,6-tetra-O-sulfonato-α-d-glucopyranosyl)-(1→4)-(β-d-glucopyranosyl-uronate)-(1→4)-2,3,6-tri-O-sulfonato-α-d-glucopyranoside] (14)
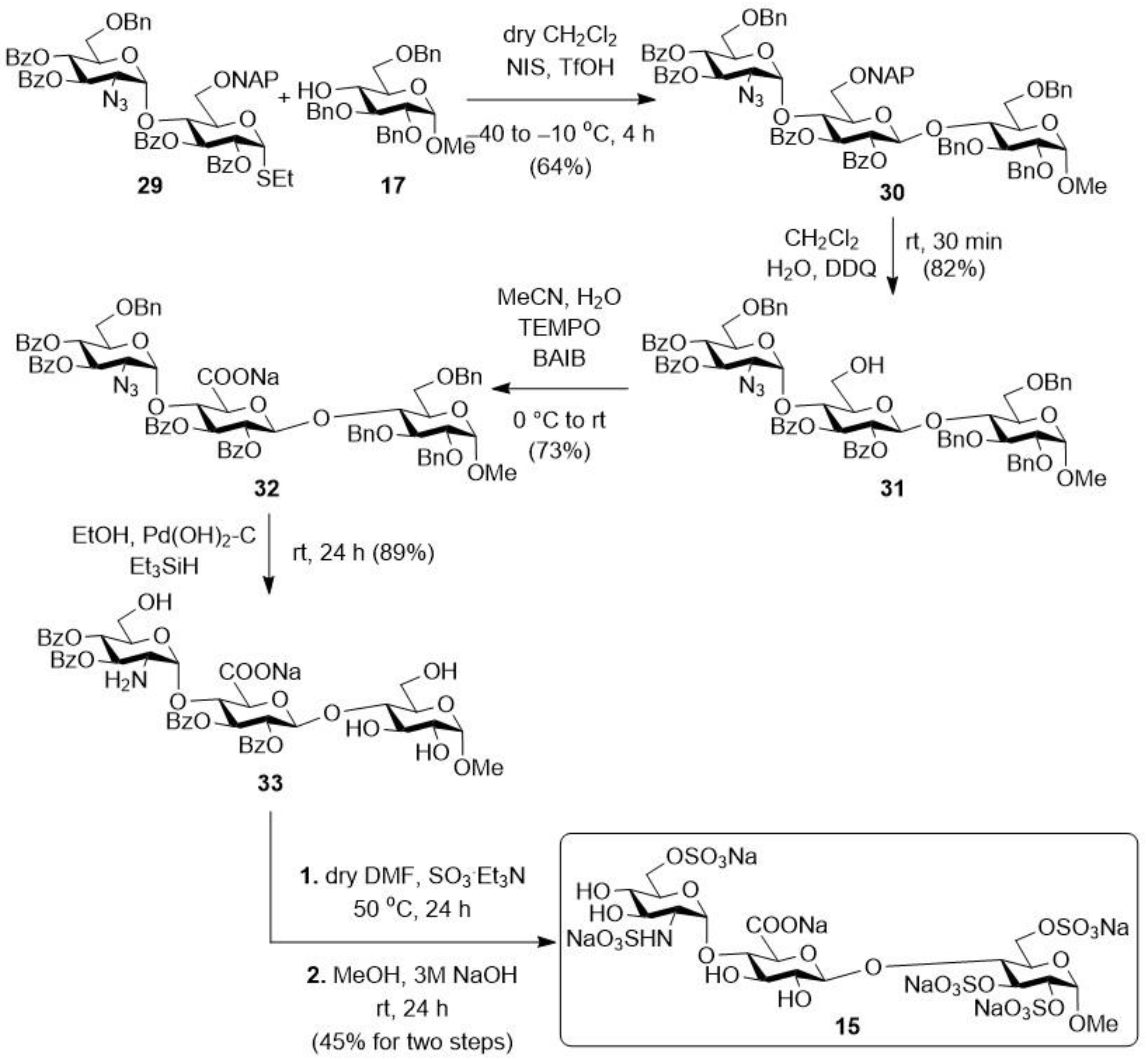
2.1.7. Hexa Sodium [methyl (2-deoxy-2-sulfamido-6-O-sulfonato-α-d-glucopyranosyl)-(1→4)-(β-d-glucopyranosyl-uronate)-(1→4)-2,3,6-tri-O-sulfonato-α-d-glucopyranoside] (15)
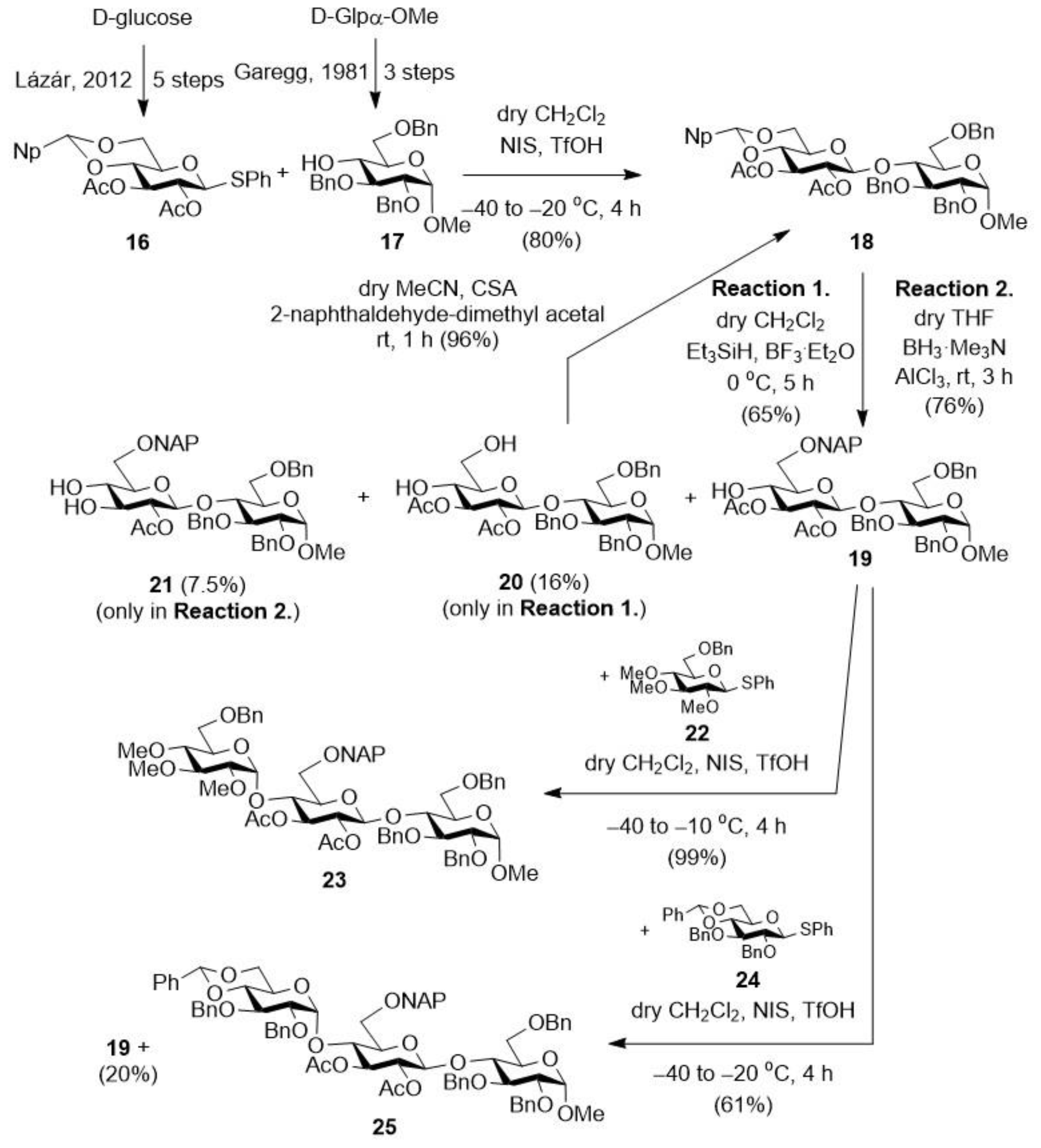
2.1.8. Methyl [2,3-di-O-acetyl-4,6-O-(2-naphthyl)methylene-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (18)
2.1.9. Methyl [2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (19), and Methyl [2,3-di-O-acetyl-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (20) and Methyl [2,3-di-O-acetyl-4-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (21)
2.1.10. Methyl (2,3-di-O-benzyl-4,6-O-benzylidene-α-d-glucopyranosyl)-(1→4)-[2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (25)
2.1.11. Methyl (2,3,6-tri-O-benzyl-α-d-glucopyranosyl)-(1→4)-[2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (26)
2.1.12. Methyl (2,3,6-tri-O-benzyl-α-d-glucopyranosyl)-(1→4)-(2,3-di-O-acetyl-β-d-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (27)
2.1.13. Sodium [methyl (2,3,6-tri-O-benzyl-α-d-glucopyranosyl)-(1→4)-(2,3-di-O-acetyl-β-d-glucopyranosyl-uronate)-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (28)
2.1.14. Methyl (2-azido-3,4-di-O-benzoyl-6-O-benzyl-2-deoxy-α-d-glucopyranosyl)-(1→4)-[2,3-di-O-benzoyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (30)
2.1.15. Methyl (2-azido-3,4-di-O-benzoyl-6-O-benzyl-2-deoxy-α-d-glucopyranosyl)-(1→4)-(2,3-di-O-benzoyl-β-d-glucopyranosyl)-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (31)
2.1.16. Sodium [methyl (2-azido-3,4-di-O-benzoyl-6-O-benzyl-2-deoxy-α-d-glucopyranosyl)-(1→4)-(2,3-di-O-benzoyl-β-d-glucopyranosyl-uronate)-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (32)
2.1.17. Sodium [methyl (2-amino-3,4-di-O-benzoyl-2-deoxy-α-d-glucopyranosyl)-(1→4)-(2,3-di-O-benzoyl-β-d-glucopyranosyl-uronate)-(1→4)-α-d-glucopyranoside (33)
2.2. Biological Investigations
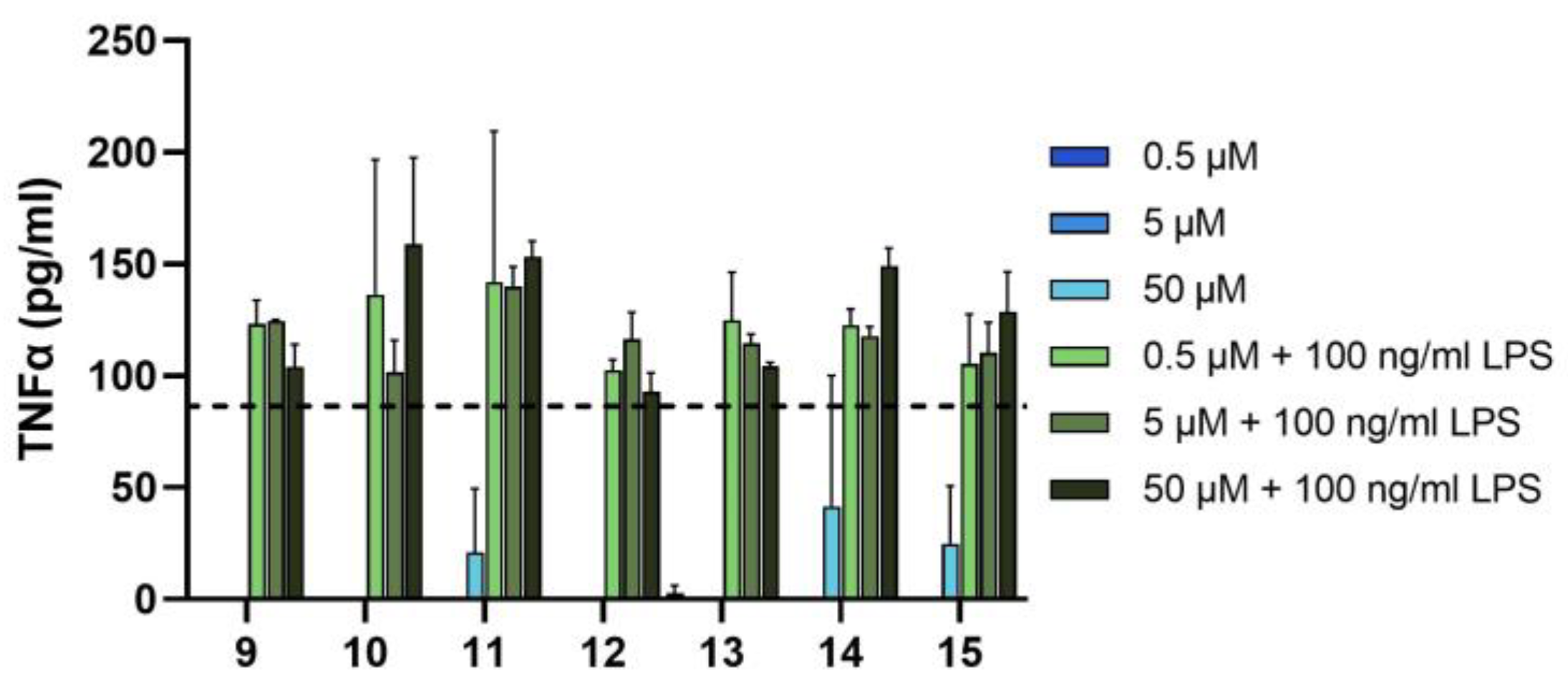
2.2.1. Investigation of the Anti-Inflammatory Effect on THP-1 Cells
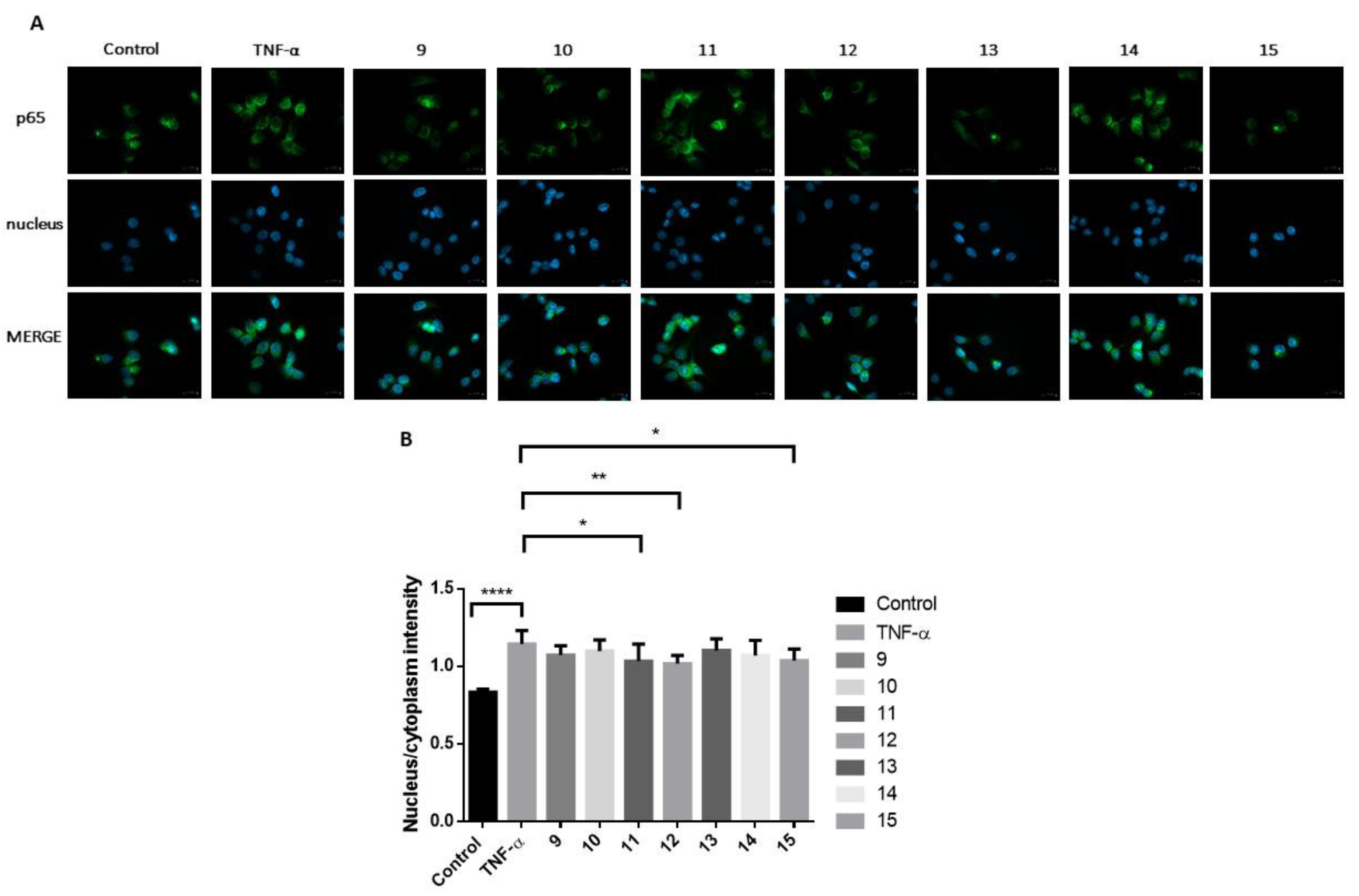
2.2.2. Investigation of the NF-κB Inflammatory Pathway
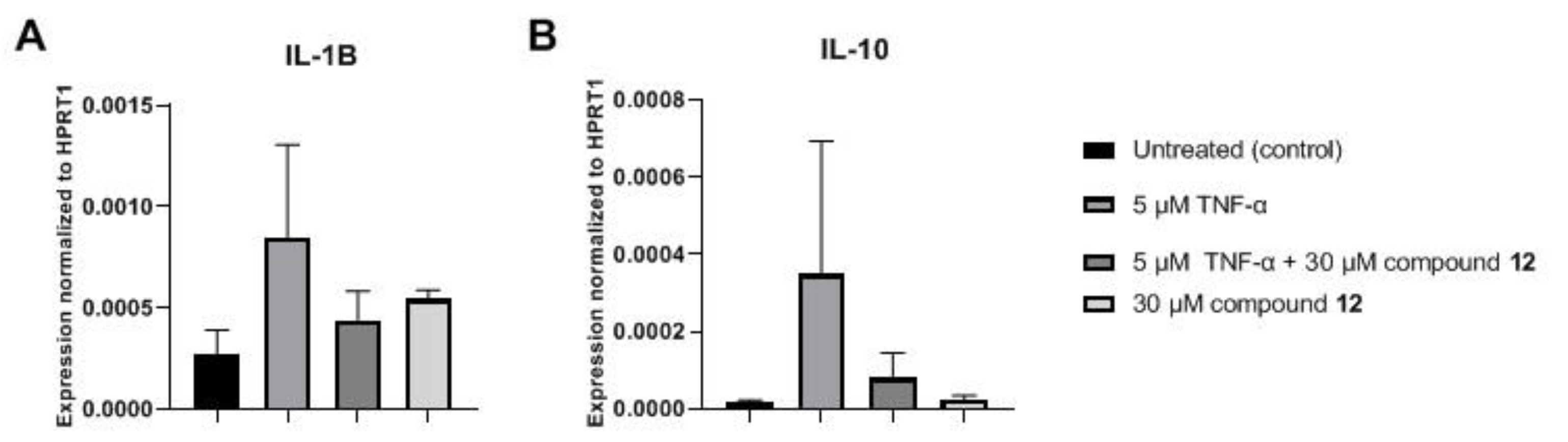
Investigation of the Inflammatory Markers Interleukin (IL)-1B and IL-10 on MCF-7 Cells
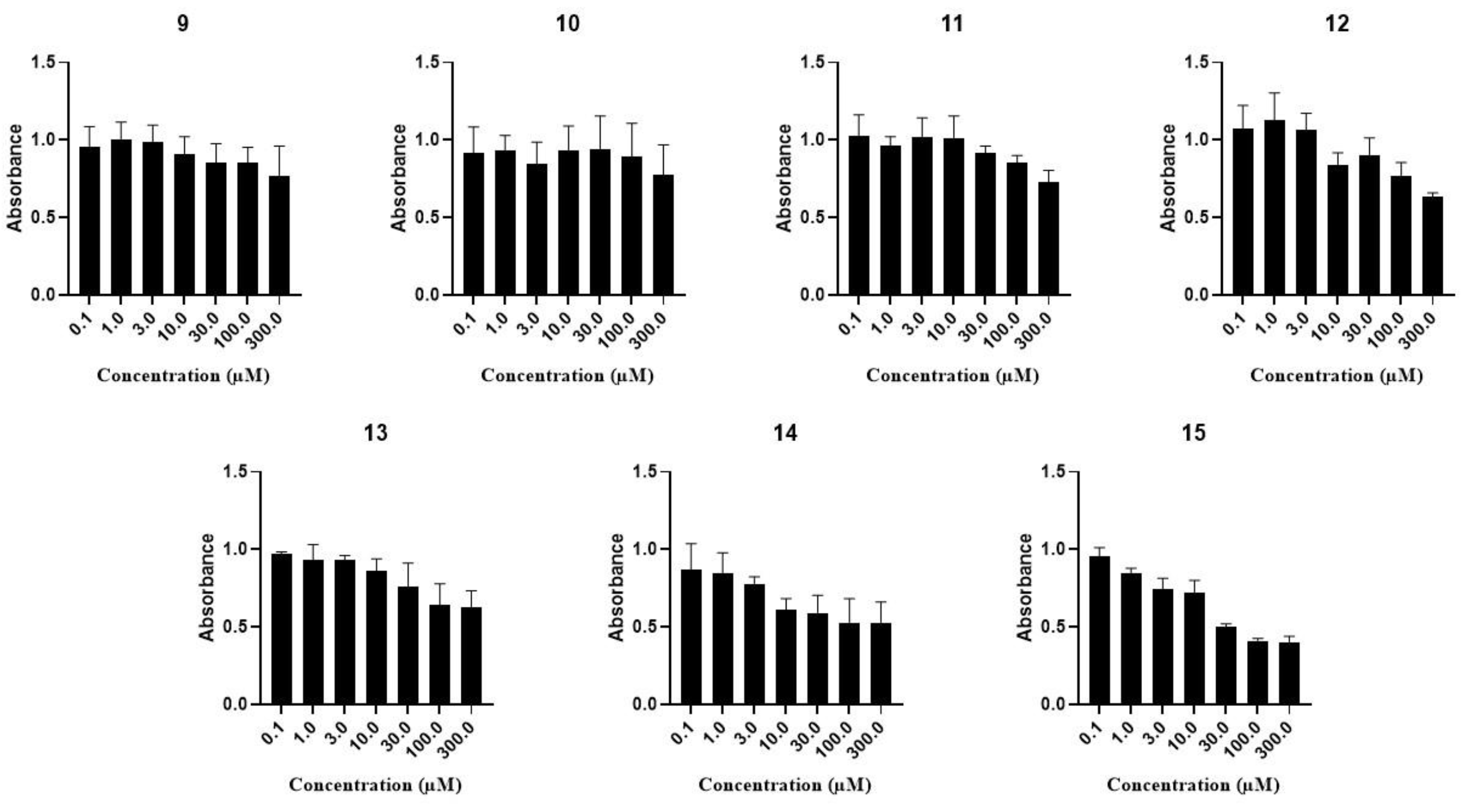
2.2.3. Investigation of the Cell Growth Inhibitory Activity on MCF-7, A2780, WM35 and HaCaT Cell Lines
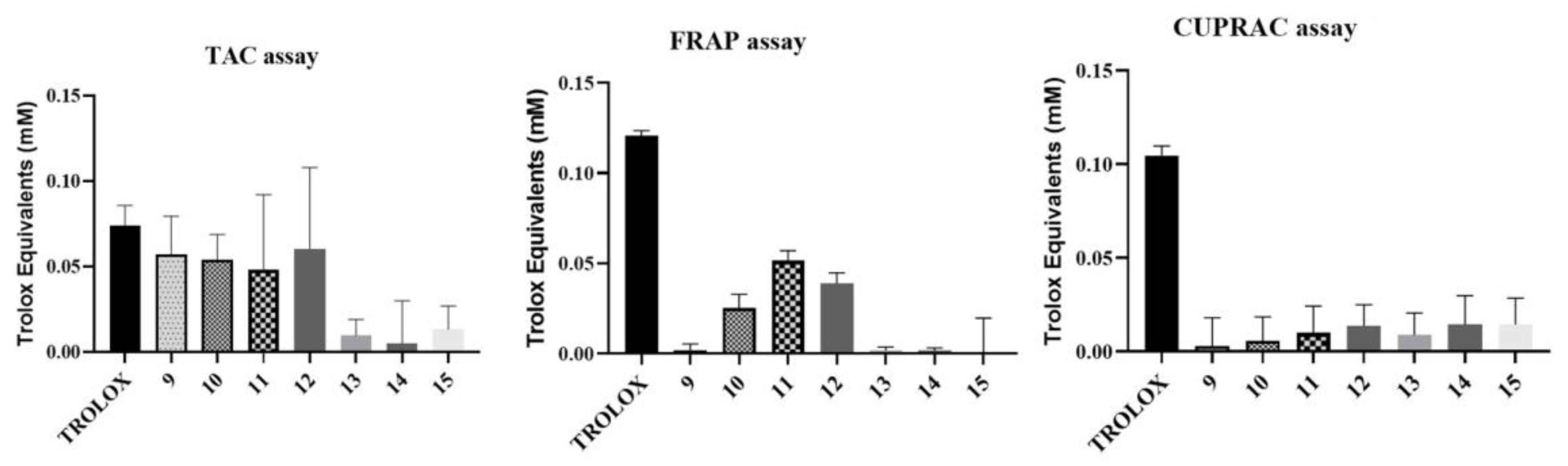
2.2.4. Investigation of the Antioxidant Activity of the Trisaccharides
TAC Assay
ORAC Assay
FRAP Assay
CUPRAC Assay
3. Results
3.1. Synthesis of the Trisaccharides
3.2. Biological Investigations
3.2.1. Investigation of the Anti-Inflammatory Effect on THP-1 Cell Line
3.2.2. Investigation of the Effect of the Trisaccharides on MCF-7 Cell Line
Investigation of the NF-κB Signalling Pathway
Investigation of the Inflammatory Markers Interleukin (IL)-1B and IL-10 on MCF-7 Cells
Cytotoxicity Studies on MCF-7 Cells
Investigation of the Cell Growth Inhibitory Activity on A2780, WM35 and HaCaT Cell Lines
3.2.3. Investigation of the Antioxidant Activity of the Trisaccharides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murata, K.; Ochiai, Y.; Akashio, K. Polydispersity of acidic glycosaminoglycan components in human liver and the changes at different stages in liver cirrhosis. Gastroenterology 1985, 89, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.L.; Busch, S.J.; Cardin, A.D. Glycosaminoglycans: Molecular properties, protein interactions, and role in physiological processes. Physiol. Rev. 1991, 71, 481–539. [Google Scholar] [CrossRef] [PubMed]
- Yeung, B.K.S.; Chong, P.Y.C.; Petillo, P.A. Synthesis of glycosaminoglycans. J. Carbohydr. Chem. 2002, 21, 799–865. [Google Scholar] [CrossRef]
- Grand, E.; Kovensky, J.; Pourceau, G.; Toumieux, S.; Wadouachi, A. Chapter 11: Anionic oligosaccharides: Synthesis and applications. In Carbohydrate Chemistry: Chemical and Biological Approaches, 1st ed.; Rauter, A.P., Lindhorst, T., Queneau, Y., Eds.; The Royal Society of Chemistry, Thomas Graham House, Science Park, Milton Road: Cambridge, UK, 2014; Volume 40, pp. 195–235. [Google Scholar]
- Mende, M.; Bednarek, C.; Wawryszyn, M.; Sauter, P.; Biskup, M.B.; Schepers, U.; Bräse, S. Chemical Synthesis of Glycosaminoglycans. Chem. Rev. 2016, 116, 8193–8255. [Google Scholar] [CrossRef]
- Jin, L.; Abrahams, J.P.; Skinner, R.; Petitou, M.; Pike, R.N.; Carrell, R.W. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. USA 1997, 94, 14683–14688. [Google Scholar] [CrossRef]
- Desai, U.R.; Petitou, M.; Bjork, I.; Olson, S.T. Mechanism of Heparin Activation of Antithrombin: Role of Individual Residues of the Pentasaccharide Activating Sequence in the Recognition of Native and Activated States of Antithrombin. J. Biol. Chem. 1998, 273, 7478–7487. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin–Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Kjelle, L.; Lindahl, U. Specificity of glycosaminoglycan-protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 101–108. [Google Scholar] [CrossRef]
- Weiss, R.J.; Esko, J.D.; Tor, Y. Targeting heparin and heparan sulfate protein interactions. Org. Biomol. Chem. 2017, 15, 5656–5668. [Google Scholar] [CrossRef]
- Cassinelli, G.; Naggi, A. Old and new applications of non-anticoagulant heparin. Int. J. Cardiol. 2016, 212S1, S14–S21. [Google Scholar] [CrossRef]
- Glantz, M.J.; Burger, P.C.; Friedman, A.H.; Radtke, R.A.; Massey, E.W.; Schold, S.C. Treatment of radiation-induced nervous system injury with heparin and warfarin. Neurology 1994, 44, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Guerrini, M.; Guglieri, S.; Naggi, A.; Perez, M.; Torri, G.; Cassinelli, G.; Ribatti, D.; Carminati, P.; Giannini, G.; et al. Undersulfated and Glycol-Split Heparins Endowed with Antiangiogenic Activity. J. Med. Chem. 2004, 47, 838–848. [Google Scholar] [CrossRef]
- Pisano, C.; Aulicino, C.; Vesci, L.; Casu, B.; Naggi, A.; Torri, G.; Ribatti, D.; Belleri, M.; Rusnati, M.; Presta, M. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic VEGF antagonist. Glycobiology 2005, 15, 1C–6C. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Bugatti, A.; Urbinati, C.; Rusnati, M. Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use. Molecules 2015, 20, 6342–6388. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J. Therapeutic use of heparin beyond anticoagulation. Curr. Drug Discov. Technol. 2009, 6, 281–289. [Google Scholar] [CrossRef]
- Zhou, H.; Roy, S.; Cochran, E.; Zouaoui, R.; Chu, C.L.; Duffner, J.; Zhao, G.; Smith, S.; Galcheva-Gargova, Z.; Karlgren, J.; et al. M402, a Novel Heparan Sulfate Mimetic, Targets Multiple Pathways Implicated in Tumor Progression and Metastasis. PLoS ONE 2011, 6, e21106. [Google Scholar] [CrossRef]
- Vogt, A.M.; Pettersson, F.; Moll, K.; Jonsson, C.; Normark, J.; Ribacke, U.; Egwang, T.G.; Ekre, H.-P.; Spillmann, D.; Chen, Q.; et al. Release of Sequestered Malaria Parasites upon Injection of a Glycosaminoglycan. PloS Pathog. 2006, 2, e100. [Google Scholar] [CrossRef]
- Kwon, P.S.; Oh, H.; Kwon, S.-J.; Jin, W.; Zhang, F.; Fraser, K.; Hong, J.J.; Linhardt, R.J.; Dordick, J.S. Sulfated polysaccharides effectively inhibit SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 50. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jin, W.; Sood, A.; Montgomery, D.W.; Grant, O.C.; Fuster, M.M.; Fu, L.; Dordick, J.S.; Woods, R.J.; Zhang, F.; et al. Characterization of heparin and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) spike glycoprotein binding interactions. Antivir. Res. 2020, 181, 104873. [Google Scholar] [CrossRef]
- Yu, Y.; Shen, M.; Song, Q.; Xie, J. Biological activities and pharmaceutical applications of polysaccharide from natural resources: A review. Carbohydr. Polym. 2018, 183, 91–101. [Google Scholar] [CrossRef]
- Xie, J.-H.; Wang, Z.-J.; Shen, M.-Y.; Nie, S.-P.; Gong, B.; Li, H.-S.; Zhao, Q.; Li, W.-J.; Xie, M.-Y. Sulfated modification, characterization and antioxidant activities of polysaccharide from Cyclocarya paliurus. Food Hydrocoll. 2016, 53, 7–15. [Google Scholar] [CrossRef]
- Shao, P.; Chen, X.; Sun, P. In vitro antioxidant and antitumor activities of different sulfated polysaccharides isolated from three algae. Int. J. Biol. Macromol. 2013, 62, 155–161. [Google Scholar] [CrossRef]
- van Boeckel, C.A.A.; Beetz, T.; Vos, J.N.; Dejong, A.J.M.; van Aelst, S.F.; van den Bosch, R.H.; Mertens, J.M.R.; van der Vlugt, F.A. Synthesis of a Pentasaccharide Corresponding to the Antithrombin III Binding Fragment of Heparin. J. Carbohydr. Chem. 1985, 4, 293–321. [Google Scholar] [CrossRef]
- van Boeckel, C.A.A.; Petitou, M. The Unique Antithrombin III Binding Domain of Heparin: A Lead to New Synthetic Antithrombotics. Angew. Chem. Int. Ed. 1993, 32, 1671–1690. [Google Scholar] [CrossRef]
- Choay, J.; Lormeau, J.-C.; Petitou, M.; Sinay, P.; Fareed, J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann. N.Y. Acad. Sci. 1981, 370, 644–649. [Google Scholar] [CrossRef]
- Petitou, M.; Duchaussoy, P.; Lederman, I.; Choay, J.; Sinay, P.; Jacquinet, J.C.; Torri, G. Synthesis of heparin fragments. A chemical synthesis of the pentasaccharide O-(2-deoxy-2-sulfamido-6-O-sulfo-α-d-glucopyranosyl) -(1→4)-O-(β-d-glucopyranosyluronic acid)-(1→4)-O-(2-deoxy-2-sulfamido-3,6-di-O-sulfo-α-d-glucopyranosyl)-(1→4)-O-(2-O-sulfo-α-l-idopyranosyluronic acid)-(1→4)-2-deoxy-2-sulfamido-6-O-sulfo-d-glucopyranose decasodium salt, a heparin fragment having high affinity for antithrombin III. Carbohydr. Res. 1986, 147, 221–236. [Google Scholar]
- Thunberg, L.; Backstrom, G.; Lindahl, U. Further characterization of the antithrombin-binding sequence in heparin. Carbohydr. Res. 1982, 100, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Lisztes, E.; Mező, E.; Demeter, F.; Horváth, L.; Bősze, S.; Tóth, B.I.; Borbás, A.; Herczeg, M. Synthesis and Cell Growth Inhibitory Activity of Six Non-glycosaminoglycan-Type Heparin-Analogue Trisaccharides. ChemMedChem 2021, 16, 1467–1476. [Google Scholar] [CrossRef]
- Peterson, S.; Liu, J. Deciphering mode of action of heparanase using structurally defined oligosaccharides. J. Biol. Chem. 2012, 287, 34836–34843. [Google Scholar] [CrossRef]
- Ni, M.; Elli, S.; Naggi, A.; Guerrini, M.; Torri, G.; Petitou, M. Investigating Glycol-Split-Heparin-Derived Inhibitors of Heparanase: A Study of Synthetic Trisaccharides. Molecules 2016, 21, 1602. [Google Scholar] [CrossRef]
- Garegg, P.J.; Hultberg, H. A novel, reductive ring-opening of carbohydrate benzylidene acetals, with unusual regioselectivity. Carbohydr. Res. 1981, 93, C10–C11. [Google Scholar] [CrossRef]
- Lázár, L.; Mező, E.; Herczeg, M.; Lipták, A.; Antus, S.; Borbás, A. Synthesis of the non-reducing end trisaccharide of the antithrombin-binding domain of heparin and its bioisosteric sulfonic acid analogues. Tetrahedron 2012, 68, 7386–7399. [Google Scholar] [CrossRef]
- Tóth, B.I.; Dobrosi, N.; Dajnoki, A.; Czifra, G.; Oláh, A.; Szöllősi, A.G.; Juhász, I.; Sugawara, K.; Paus, R.; Bíró, T. Endocannabinoids modulate human epidermal keratinocyte proliferation and survival via the sequential engagement of cannabinoid receptor-1 and transient receptor potential vanilloid-1. J. Invest. Dermatol. 2011, 131, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Szőke, K.; Kajtár, R.; Gyöngyösi, A.; Czompa, A.; Fésüs, A.; Lőrincz, E.B.; Petróczi, F.D.; Herczegh, P.; Bak, I.; Borbás, A.; et al. Pharmacological Evaluation of Newly Synthesized Cannabidiol Derivates on H9c2 Cells. Antioxidants 2023, 12, 1714. [Google Scholar] [CrossRef]
- DeLange, R.J.; Glazer, A.N. Phycoerythrin fluorescence-based assay for peroxy radicals: A screen for biologically relevant protective agents. Analyt Biochem. 1989, 177, 300–306. [Google Scholar] [CrossRef]
- Csepanyi, E.; Szabados-Furjesi, P.; Kiss-Szikszai, A.; Frensemeier, L.M.; Karst, U.; Lekli, I.; Haines, D.D.; Tosaki, A.; Bak, I. Antioxidant Properties and Oxidative Transformation of Different Chromone Derivatives. Molecules 2017, 22, 588. [Google Scholar] [CrossRef] [PubMed]
- Apak, R.; Güclü, K.; Özyürek, M.; Karademir, S.E. Novel Total Antioxidant Capacity Index for Dietary Polyphenols and Vitamins C and E, Using Their Cupric Ion Reducing Capability in the Presence of Neocuproine: CUPRAC Method. J. Agric. Food Chem. 2004, 52, 7970–7981. [Google Scholar] [CrossRef]
- Debenham, S.D.; Toone, E.J. Regioselective reduction of 4,6-O-benzylidenes using triethylsilane and BF3·Et2O Tetrahedron. Asymmetry 2000, 11, 385–387. [Google Scholar] [CrossRef]
- Ek, M.; Garegg, P.J.; Hultberg, H.; Oscarson, S. Reductive Ring Openings of Carbohydrate Benzylidene Acetals Using Borane-Trimethylamine and Aluminium Chloride. Regioselectivity and Solvent Dependance. J. Carbohydr. Chem. 1983, 2, 305–311. [Google Scholar] [CrossRef]
- Ohlin, M.; Johnsson, R.; Ellervik, U. Regioselective reductive openings of 4,6-benzylidene acetals: Synthetic and mechanistic aspects. Carbohydr. Res. 2011, 346, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Abbas, S.A.; Locke, R.D.; Piskorz, C.F.; Alderfer, J.L.; Matta, K.L. Use of 1,2-dichloro 4,5-dicyanoquinone (DDQ) for cleavage of the 2-naphthylmethyl (NAP) group. Tetrahedron Lett. 2000, 41, 169–173. [Google Scholar] [CrossRef]
- Despras, G.; Bernard, C.; Perrot, A.; Cattiaux, L.; Prochiantz, A.; Lortat-Jacob, H.; Mallet, J.-M. Toward Libraries of Biotinylated Chondroitin Sulfate Analogues: From Synthesis to In Vivo Studies. Chem. Eur. J. 2013, 19, 531–540. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demeter, F.; Peleskei, Z.; Kútvölgyi, K.; Rusznyák, Á.; Fenyvesi, F.; Kajtár, R.; Sipos, É.; Lekli, I.; Molnár, P.; Szöllősi, A.G.; et al. Synthesis and Biological Profiling of Seven Heparin and Heparan Sulphate Analogue Trisaccharides. Biomolecules 2024, 14, 1052. https://doi.org/10.3390/biom14091052
Demeter F, Peleskei Z, Kútvölgyi K, Rusznyák Á, Fenyvesi F, Kajtár R, Sipos É, Lekli I, Molnár P, Szöllősi AG, et al. Synthesis and Biological Profiling of Seven Heparin and Heparan Sulphate Analogue Trisaccharides. Biomolecules. 2024; 14(9):1052. https://doi.org/10.3390/biom14091052
Chicago/Turabian StyleDemeter, Fruzsina, Zsófia Peleskei, Katalin Kútvölgyi, Ágnes Rusznyák, Ferenc Fenyvesi, Richárd Kajtár, Éva Sipos, István Lekli, Petra Molnár, Attila Gábor Szöllősi, and et al. 2024. "Synthesis and Biological Profiling of Seven Heparin and Heparan Sulphate Analogue Trisaccharides" Biomolecules 14, no. 9: 1052. https://doi.org/10.3390/biom14091052
APA StyleDemeter, F., Peleskei, Z., Kútvölgyi, K., Rusznyák, Á., Fenyvesi, F., Kajtár, R., Sipos, É., Lekli, I., Molnár, P., Szöllősi, A. G., Lisztes, E., Tóth, B. I., Borbás, A., & Herczeg, M. (2024). Synthesis and Biological Profiling of Seven Heparin and Heparan Sulphate Analogue Trisaccharides. Biomolecules, 14(9), 1052. https://doi.org/10.3390/biom14091052