
Biophysical and Solution Structure Analysis of Critical Residues Involved in the Interaction between the PupB N-Terminal Signaling Domain and PupR C-Terminal Cell Surface Signaling Domain from Pseudomonas capeferrum
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of the GST-PupB NTSD and MBP-PupR CCSSD and Mutagenesis
2.2. Expression and Purification of PupB NTSD and Mutants
2.3. Quantification of the MBP-PupR CCSSD Interaction with PupB NTSD Mutants
2.4. CD Spectroscopy and Thermal Denaturation of PupB NTSD Mutants
2.5. NMR Spectroscopy
2.5.1. NMR Sample Preparation, Data Collection, and Chemical Shift Assignment
2.5.2. NMR Hydrogen/Deuterium Exchange Experiments
2.5.3. Hydrogen Bond Restraints
2.5.4. Dihedral Angle Restraints
2.5.5. NOE Assignment and Structure Calculation
2.5.6. Structure Validation
2.5.7. 1H–15N HSQC Spectroscopy of Labelled PupB NTSD Mutants
3. Results
3.1. Solution NMR Structure of the PupB NTSD
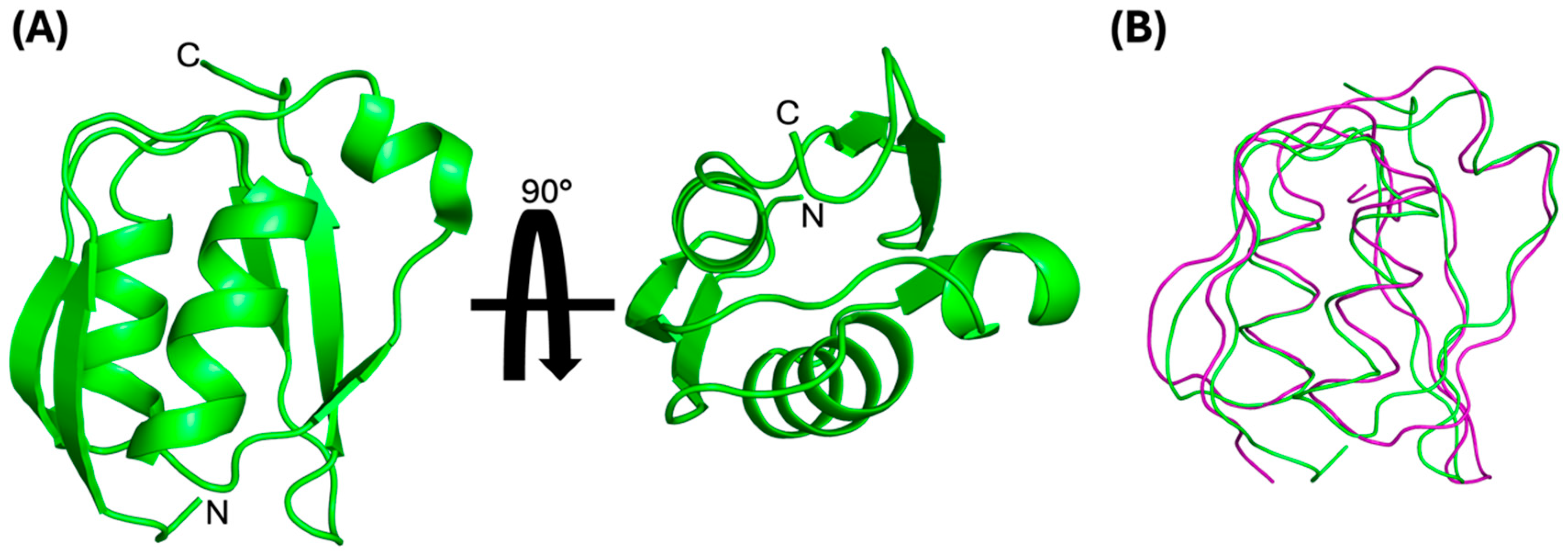
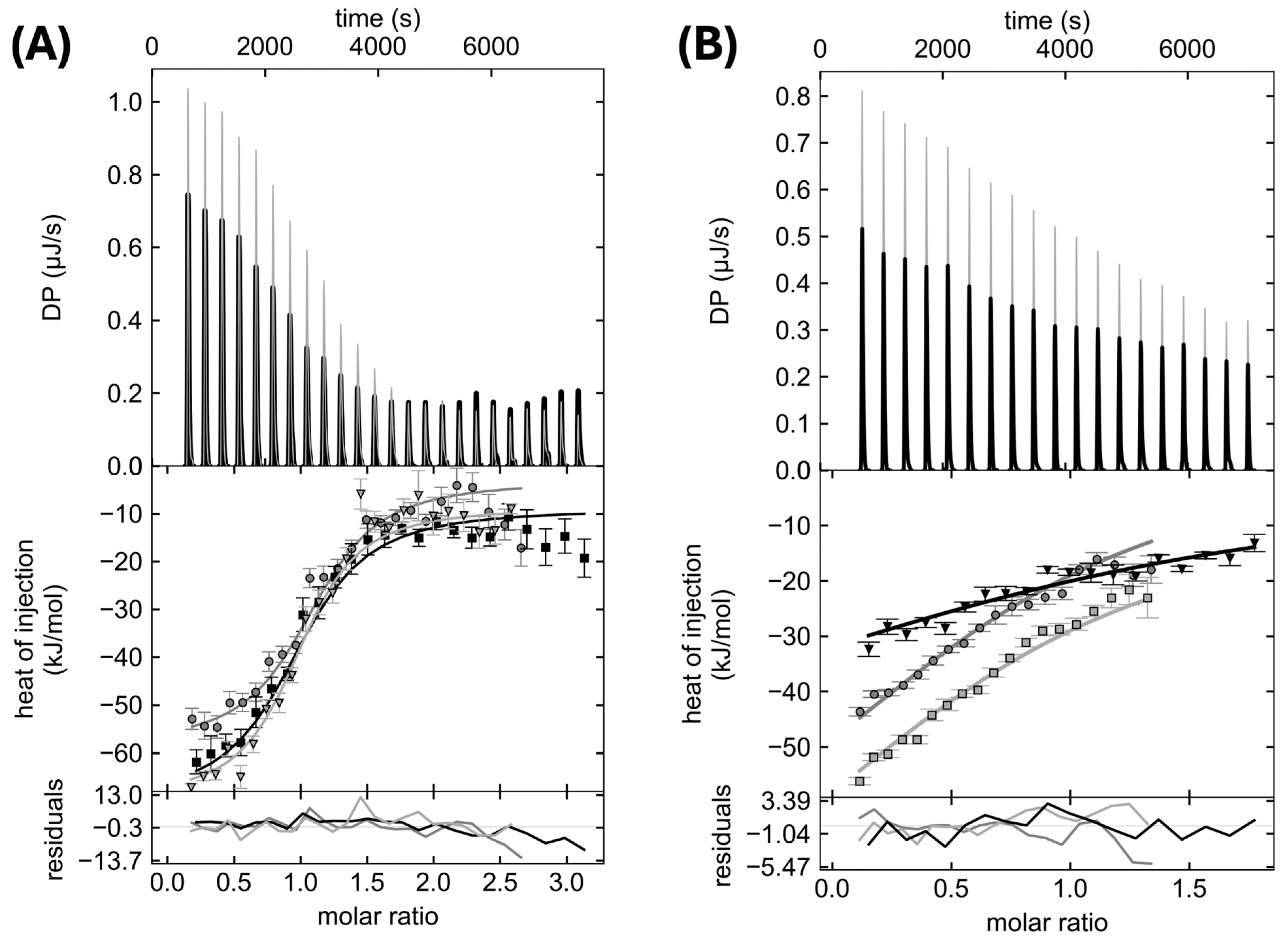
3.2. Effect of Mutations on Interaction between the NTSD and the CCSSD
3.3. Effect of NTSD Mutations on Folding and Thermal Stability
3.4. Comparison of 1H–15N HSQC Mutant and WT Spectra Indicates the Structural Effects of Mutation Are Modest
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Cell surface signaling | CSS |
| C-terminal cell surface signaling domain | CCSSD |
| Circular dichroism spectroscopy | CD |
| Ferric citrate | Fec |
| Ferric pyoverdine | Fpv |
| Isothermal titration calorimetry | ITC |
| N-terminal signaling domain | NTSD |
| Nuclear magnetic resonance | NMR |
| Pseudobactin BN7/8 uptake | Pup |
| Wild-type | WT |
References
- Ferguson, A.D.; Deisenhofer, J. Metal Import through Microbial Membranes. Cell 2004, 116, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Guillier, M.; Barnard, T.J.; Buchanan, S.K. TonB-Dependent Transporters: Regulation, Structure, and Function. Annu. Rev. Microbiol. 2010, 64, 43–60. [Google Scholar] [CrossRef]
- Llamas, M.A.; Imperi, F.; Visca, P.; Lamont, I.L. Cell-surface signaling in Pseudomonas: Stress responses, iron transport, and pathogenicity. FEMS Microbiol. Rev. 2014, 38, 569–597. [Google Scholar] [CrossRef]
- Koster, M.; van de Vossenberg, J.; Leong, J.; Weisbeek, P.J. Identification and characterization of the pupB gene encoding an inducible ferric-pseudobactin receptor of Pseudomonas putida WCS358. Mol. Microbiol. 1993, 8, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.; van Klompenburg, W.; Bitter, W.; Leong, J.; Weisbeek, P. Role for the outer membrane ferric siderophore receptor PupB in signal transduction across the bacterial cell envelope. Embo J. 1994, 13, 2805–2813. [Google Scholar] [CrossRef]
- Van Hove, B.; Staudenmaier, H.; Braun, V. Novel two-component transmembrane transcription control: Regulation of iron dicitrate transport in Escherichia coli K-12. J. Bacteriol. 1990, 172, 6749–6758. [Google Scholar] [CrossRef] [PubMed]
- Harle, C.; Kim, I.; Angerer, A.; Braun, V. Signal transfer through three compartments: Transcription initiation of the Escherichia coli ferric citrate transport system from the cell surface. EMBO J. 1995, 14, 1430–1438. [Google Scholar] [CrossRef]
- Enz, S.; Mahren, S.; Stroeher, U.H.; Braun, V. Surface Signaling in Ferric Citrate Transport Gene Induction: Interaction of the FecA, FecR, and FecI Regulatory Proteins. J. Bacteriol. 2000, 182, 637–646. [Google Scholar] [CrossRef]
- Wirth, C.; Meyer-Klaucke, W.; Pattus, F.; Cobessi, D. From the Periplasmic Signaling Domain to the Extracellular Face of an Outer Membrane Signal Transducer of Pseudomonas aeruginosa: Crystal Structure of the Ferric Pyoverdine Outer Membrane Receptor. J. Mol. Biol. 2007, 368, 398–406. [Google Scholar] [CrossRef]
- Edgar, R.J.; Xu, X.; Shirley, M.; Konings, A.F.; Martin, L.W.; Ackerley, D.F.; Lamont, I.L. Interactions between an anti-sigma protein and two sigma factors that regulate the pyoverdine signaling pathway in Pseudomonas aeruginosa. BMC Microbiol. 2014, 14, 287. [Google Scholar] [CrossRef]
- Garcia-Herrero, A.; Vogel, H.J. Nuclear magnetic resonance solution structure of the periplasmic signalling domain of the TonB-dependent outer membrane transporter FecA from Escherichia coli. Mol. Microbiol. 2005, 58, 1226–1237. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Amezcua, C.A.; Halabi, N.M.; Chelliah, Y.; Rosen, M.K.; Ranganathan, R.; Deisenhofer, J. Signal transduction pathway of TonB-dependent transporters. Proc. Natl. Acad. Sci. USA 2007, 104, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Cobessi, D.; Celia, H.; Folschweiller, N.; Schalk, I.J.; Abdallah, M.A.; Pattus, F. The Crystal Structure of the Pyoverdine Outer Membrane Receptor FpvA from Pseudomonas aeruginosa at 3.6†A Resolution. J. Mol. Biol. 2005, 347, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Brillet, K.; Journet, L.; CÈlia, H.; Paulus, L.; Stahl, A.; Pattus, F.; Cobessi, D. A [beta] Strand Lock Exchange for Signal Transduction in TonB-Dependent Transducers on the Basis of a Common Structural Motif. Structure 2007, 15, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Nader, M.; Celia, H.; Gruffaz, C.; Geoffroy, V.; Meyer, J.M.; Schalk, I.J.; Pattus, F. FpvA bound to non-cognate pyoverdines: Molecular basis of siderophore recognition by an iron transporter. Mol. Microbiol. 2009, 72, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.L.; Jernberg, B.D.; Sinha, S.C.; Colbert, C.L. Structural basis of cell-surface signaling by a conserved sigma regulator in Gram-negative bacteria. J. Biol. Chem. 2020, 295, 5795–5806. [Google Scholar] [CrossRef]
- Enz, S.; Brand, H.; Orellana, C.; Mahren, S.; Braun, V. Sites of interaction between the FecA and FecR signal transduction proteins of ferric citrate transport in Escherichia coli K-12. J. Bacteriol. 2003, 185, 3745–3752. [Google Scholar] [CrossRef]
- Jensen, J.L.; Wu, Q.; Colbert, C.L. NMR assignments of the N-terminal signaling domain of the TonB-dependent outer membrane transducer PupB. Biomol. NMR Assign. 2018, 12, 91–94. [Google Scholar] [CrossRef]
- Jensen, J.L.; Balbo, A.; Neau, D.B.; Chakravarthy, S.; Zhao, H.; Sinha, S.C.; Colbert, C.L. Mechanistic Implications of the Unique Structural Features and Dimerization of the Cytoplasmic Domain of the Pseudomonas Sigma Regulator, PupR. Biochemistry 2015, 54, 5867–5877. [Google Scholar] [CrossRef]
- Keller, S.; Vargas, C.; Zhao, H.; Piszczek, G.; Brautigam, C.A.; Schuck, P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 2012, 84, 5066–5073. [Google Scholar] [CrossRef]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef]
- Brautigam, C.A. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 2015, 562, 109–133. [Google Scholar] [PubMed]
- Brautigam, C.A.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 2016, 11, 882–894. [Google Scholar] [CrossRef]
- Sreerama, N.; Venyaminov, S.Y.; Woody, R.W. Analysis of protein circular dichroism spectra based on the tertiary structure classification. Anal. Biochem. 2001, 299, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Blevins, R.A. NMRview: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A. From Raw Data to Protein Backbone Chemical Shifts Using NMRFx Processing and NMRViewJ Analysis. Methods Mol. Biol. 2018, 1688, 257–310. [Google Scholar] [PubMed]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Bax, A.; Ikura, M.; Kay, L.E.; Barbato, G.; Spera, S. Multidimensional triple resonance NMR spectroscopy of isotopically uniformly enriched proteins: A powerful new strategy for structure determination. Ciba Found. Symp. 1991, 161, 108–119; discussion 119–135. [Google Scholar]
- Metzler, W.J.; Constantine, K.L.; Friedrichs, M.S.; Bell, A.J.; Ernst, E.G.; Lavoie, T.B.; Mueller, L. Characterization of the three-dimensional solution structure of human profilin: 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry 1993, 32, 13818–13829. [Google Scholar] [CrossRef]
- Cavanagh, J. Protein NMR Spectroscopy: Principles and Practice; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Delaglio, F.; Grzesiek, S.; Vuister, G.; Zu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Maciejewski, M.W.; Schuyler, A.D.; Gryk, M.R.; Moraru, I.I.; Romero, P.R.; Ulrich, E.L.; Eghbalnia, H.R.; Livny, M.; Delaglio, F.; Hoch, J.C. NMRbox: A Resource for Biomolecular NMR Computation. Biophys. J. 2017, 112, 1529–1534. [Google Scholar] [CrossRef]
- Rubinson, K.A. Practical corrections for p(H,D) measurements in mixed H2O/D2O biological buffers. Anal. Methods 2017, 9, 2744–2750. [Google Scholar] [CrossRef]
- Cheung, M.S.; Maguire, M.L.; Stevens, T.J.; Broadhurst, R.W. DANGLE: A Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J. Magn. Reson. 2010, 202, 223–233. [Google Scholar] [CrossRef]
- Rieping, W.; Habeck, M.; Bardiaux, B.; Bernard, A.; Malliavin, T.E.; Nilges, M. ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 2007, 23, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Huang, Y.; Baxter, R.; Smith, B.S.; Partch, C.L.; Colbert, C.L.; Deisenhofer, J. Crystal structure of cryptochrome 3 from Arabidopsis thaliana and its implications for photolyase activity. Proc. Natl. Acad. Sci. USA 2006, 103, 17701–17706. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System; Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| List of Constraints | |
|---|---|
| NOE Distance Restraints: | |
| Unambiguous | 1312 |
| Ambiguous | 181 |
| Hydrogen Bond Restraints | 50 |
| Dihedral Angle Restraints | 144 |
| Structural Analysis | |
| Mean r.m.s.d. from experimental restraints: | |
| NOE (Å) | 0.501 ± 0.031 |
| Dihedral angles (deg.) | 5.834 ± 0.927 |
| Average number of: | |
| NOE violations > 0.5 Å | 70.3 ± 2.4 |
| NOE violations > 0.3 Å | 99.9 ± 4.3 |
| Dihedral violations > 5° | 12.4 ± 1.7 |
| Mean rms from idealized covalent geometry: | |
| Bonds, Å | 0.0099 ± 0.0004 |
| Angles, ° | 1.06 ± 0.03 |
| Impropers, ° | 1.28 ± 0.06 |
| Geometric analysis of residues 2–82 | |
| Rmsd to mean, backbone, Å | 0.79 ± 0.35 |
| Rmsd to mean, all heavy, Å | 1.46 ± 0.34 |
| Ramachandran Analysis (Procheck) | |
| Most-favored region (%) | 62.5 |
| Additionally Allowed region (%) | 33.4 |
| Generously Allowed region (%) | 3.9 |
| Disallowed region (%) | 0.15 |
| PupB NTSD | Kd (µM) | ΔH (kJ/mol) | ΔS (J/mol·K) | ΔG (kJ/mol) | Incompetent Fraction (%) |
|---|---|---|---|---|---|
| WT | 0.69 [0.42, 1.11] a | −73.99 [−80.99, −68.27] a | −138.83 [−158.94, −122.93] a | −33.99 [−35.18, −32.84] a | 0.0–12.8% |
| Q69K | 1.55 [0.9, 2.6] a | −60.70 [−67.83, −55.03] a | −99.42 [−119.81, −84.05] a | −32.05 [−33.31, −30.81] a | 0.0–9.3% |
| H72D | 27.71 [U b, 125.63] a | −96.16 [U b, −67.65] a | −246.46 [U b, −122.93] a | −25.14 [U b, −21.52] a | 0.0–43.5% |
| L74A | No Interaction Detected | - | - | - | - |
| PupB NTSD | Helix | Strand | Coil + Turn | Total | Tm (Heating) | Tm (Cooling) |
|---|---|---|---|---|---|---|
| WT | 19.5 ± 1.2 | 36.6 ± 1.2 | 43.9 ± 0 | 100 | 57.4 ± 0.1 °C | 53.8 ± 0.9 °C |
| Q69K | 12.2 ± 3.7 | 37.8 ± 4.9 | 50.0 ± 1.2 | 100 | 57.8 ± 0.9 °C | 58.9 ± 0.1 °C |
| H72D | 11.0 ± 3.7 | 37.8 ± 2.4 | 51.2 ± 1.2 | 100 | 53.1 ± 0.3 °C | 53.3 ± 0.3 °C |
| L74A | 20.7 ± 1.2 | 36.6 ± 1.2 | 42.7 ± 1.2 | 100 | 56.3 ± 0.2 °C | 56.3 ± 0.2 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultana, T.; Morgan, D.M.; Jernberg, B.D.; Zak, P.; Sinha, S.C.; Colbert, C.L. Biophysical and Solution Structure Analysis of Critical Residues Involved in the Interaction between the PupB N-Terminal Signaling Domain and PupR C-Terminal Cell Surface Signaling Domain from Pseudomonas capeferrum. Biomolecules 2024, 14, 1108. https://doi.org/10.3390/biom14091108
Sultana T, Morgan DM, Jernberg BD, Zak P, Sinha SC, Colbert CL. Biophysical and Solution Structure Analysis of Critical Residues Involved in the Interaction between the PupB N-Terminal Signaling Domain and PupR C-Terminal Cell Surface Signaling Domain from Pseudomonas capeferrum. Biomolecules. 2024; 14(9):1108. https://doi.org/10.3390/biom14091108
Chicago/Turabian StyleSultana, Tajnin, David M. Morgan, Beau D. Jernberg, Peyton Zak, Sangita C. Sinha, and Christopher L. Colbert. 2024. "Biophysical and Solution Structure Analysis of Critical Residues Involved in the Interaction between the PupB N-Terminal Signaling Domain and PupR C-Terminal Cell Surface Signaling Domain from Pseudomonas capeferrum" Biomolecules 14, no. 9: 1108. https://doi.org/10.3390/biom14091108