
Attenuation of PM2.5-Induced Lung Injury by 4-Phenylbutyric Acid: Maintenance of [Ca2+]i Stability between Endoplasmic Reticulum and Mitochondria

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PM2.5 Sample Preparation
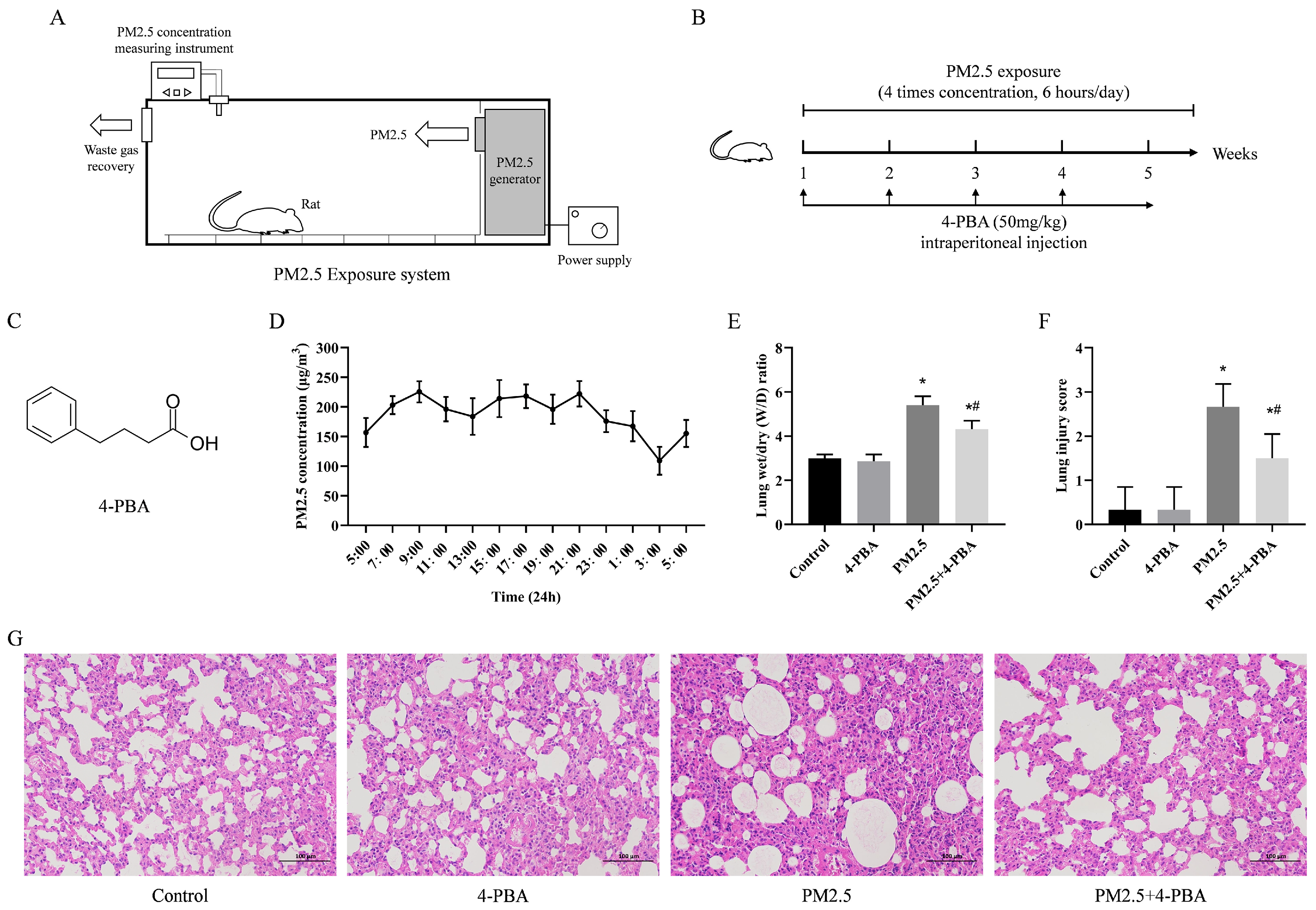
2.3. Animal Model Establishment and Grouping
2.4. Cell Model Establishment
2.5. Histopathological Examination
2.6. TUNEL Assay
2.7. Cell Viability
2.8. Cell ATP Assay
2.9. Cytoplasmic Ca2+, JC-1 and Mito-SOX Measurement
2.10. Transmission Electron Microscopy (TEM)
2.11. Immunofluorescence Staining
2.12. ELISA
2.13. RT-qPCR
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
| ATF6 | TTTGGATTTGATGCCTTGGGAGTC | CTGTGGACCGAGGAGAAGAGAC |
| CHOP | CCTCGCTCTCAAGATTCCAGTC | TCATTCTCCTGCTCCTTCTCCTTC |
| GRP78 | GGAGGAGGACAAGAAGGAGGATG | TTGAATACACCGACGCAGGAATAG |
| GAPDH | CCTGCACCACCAACTGCTTA | CATCACGCCACAGCTTTCCA |
2.14. Western Blotting
2.15. Statistical Analysis
3. Results
3.1. 4-PBA Inhibits PM2.5-Induced Lung Injury in Rats
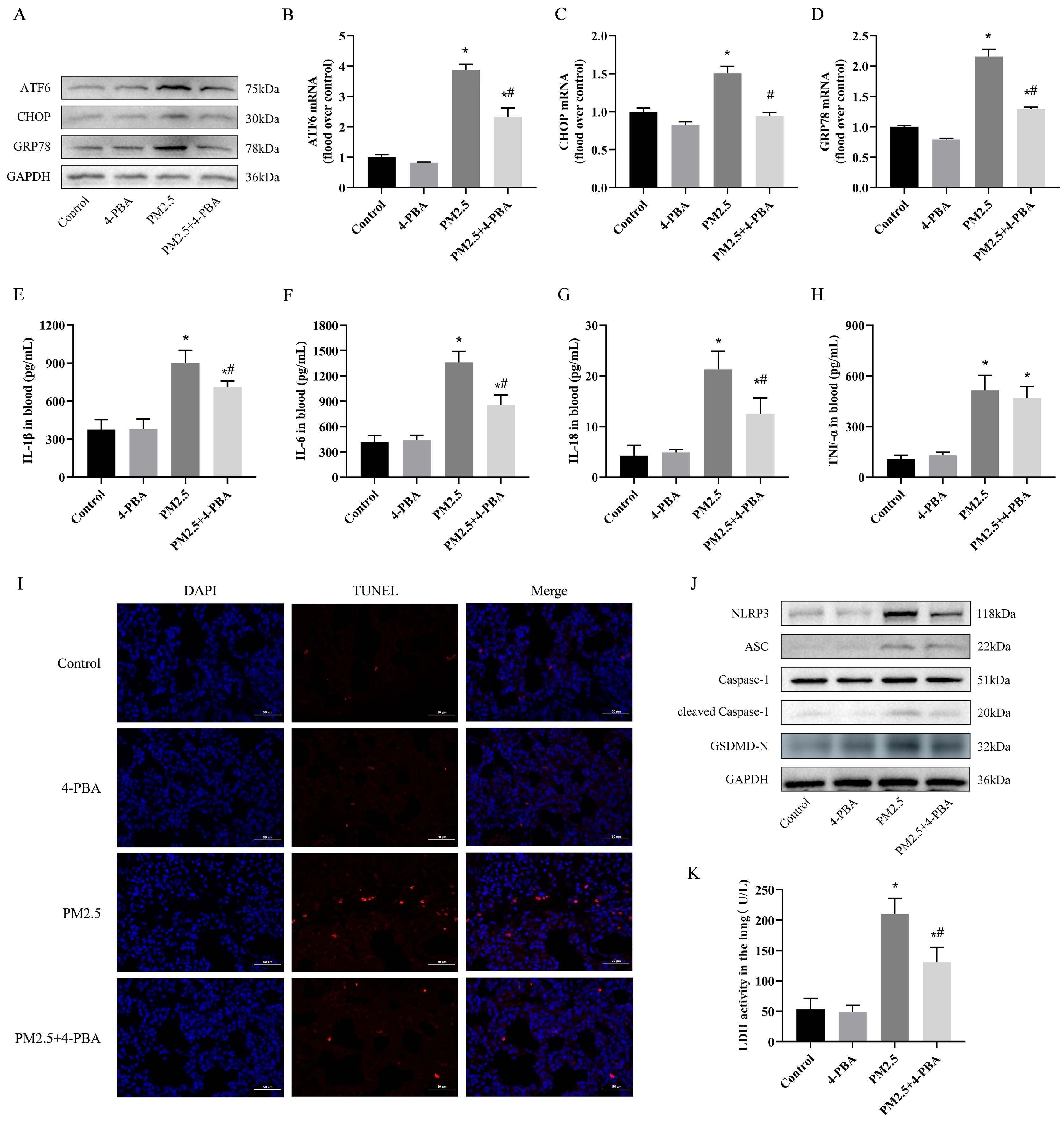
3.2. 4-PBA Inhibits PM2.5 Exposure-Induced ER Stress in Rat Lungs
3.3. 4-PBA Inhibits Lung Inflammation and Pyroptosis Induced by PM2.5 in Rats
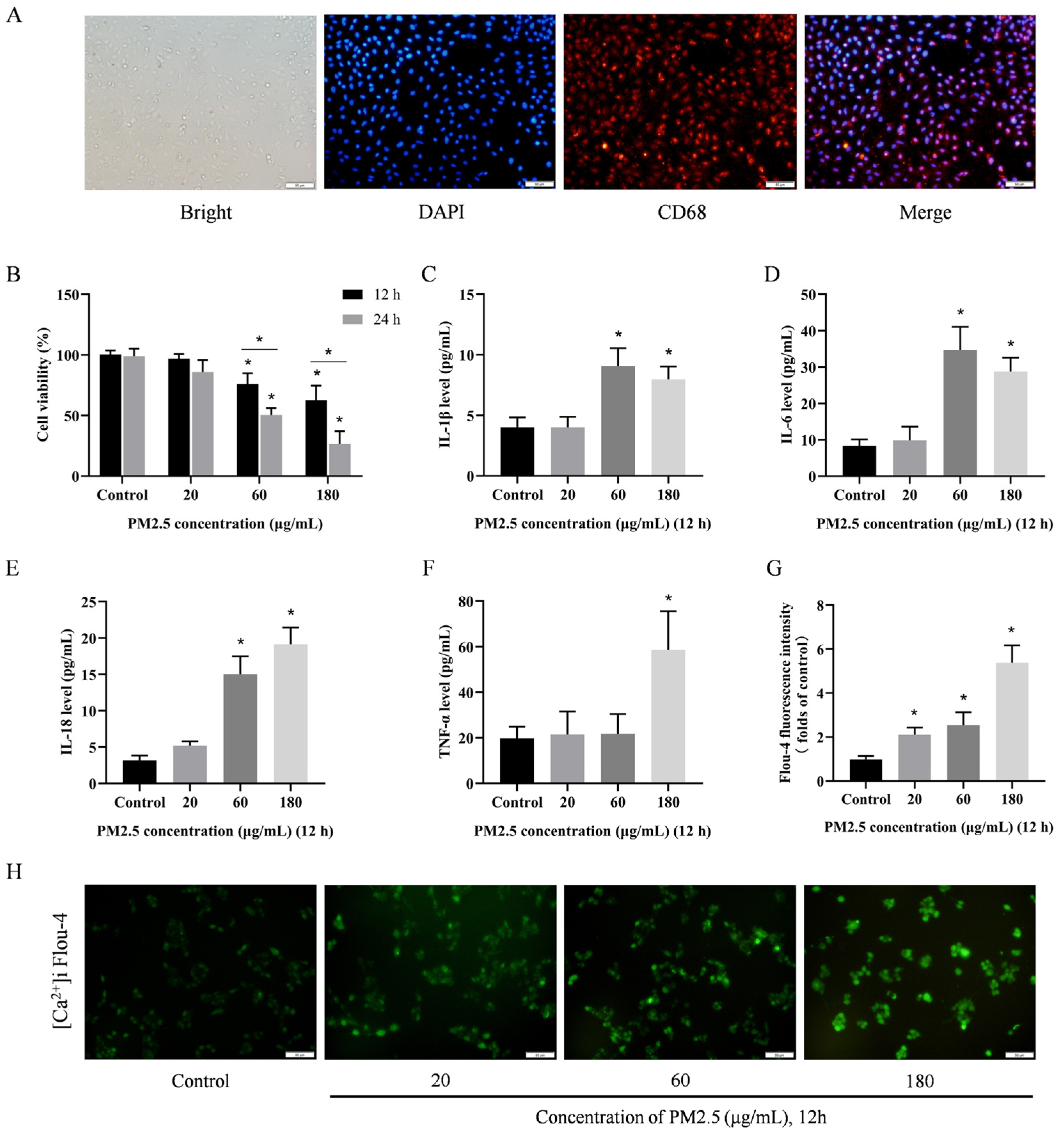
3.4. PM2.5 Induces Damage to Rat Alveolar Macrophages In Vitro
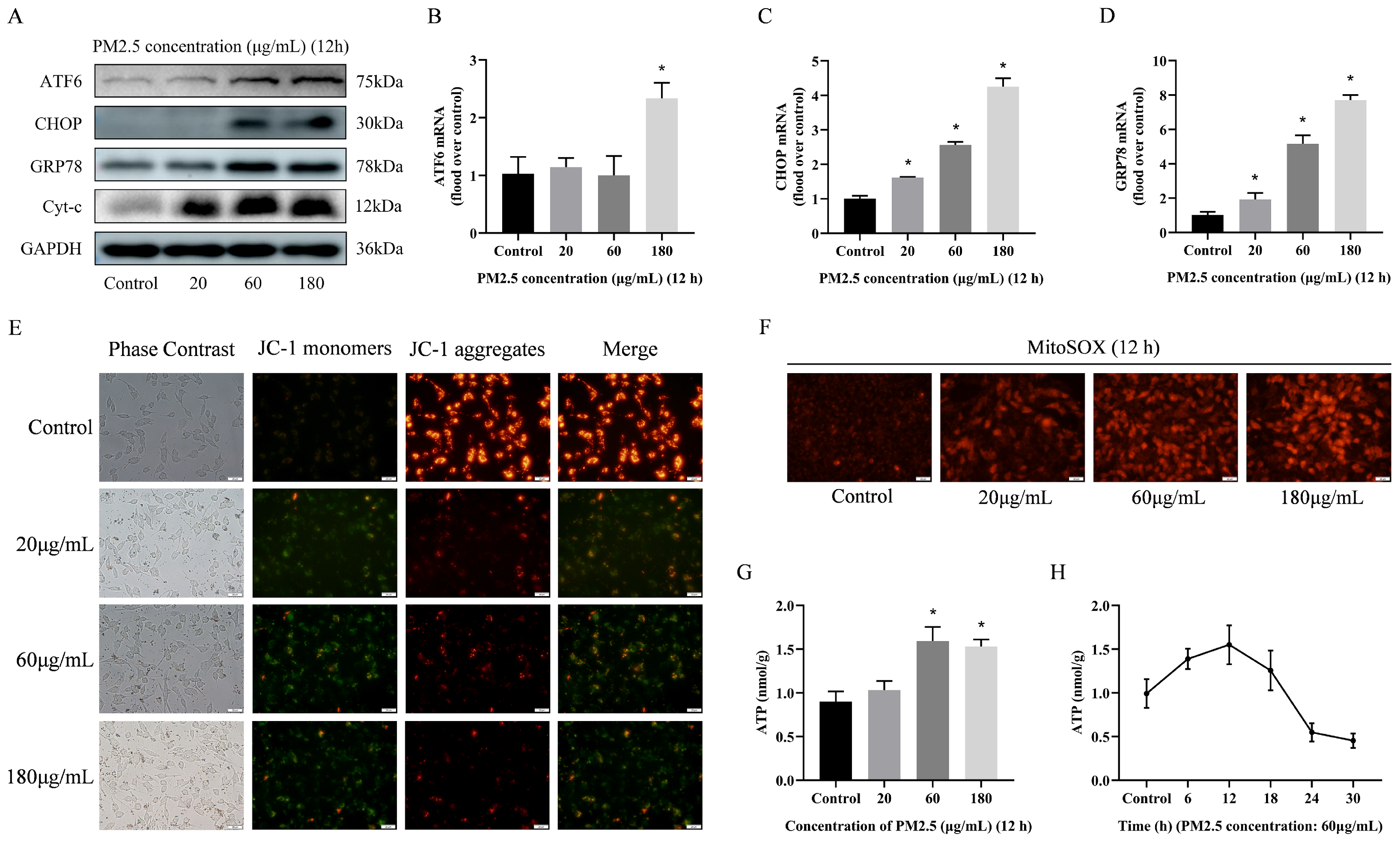
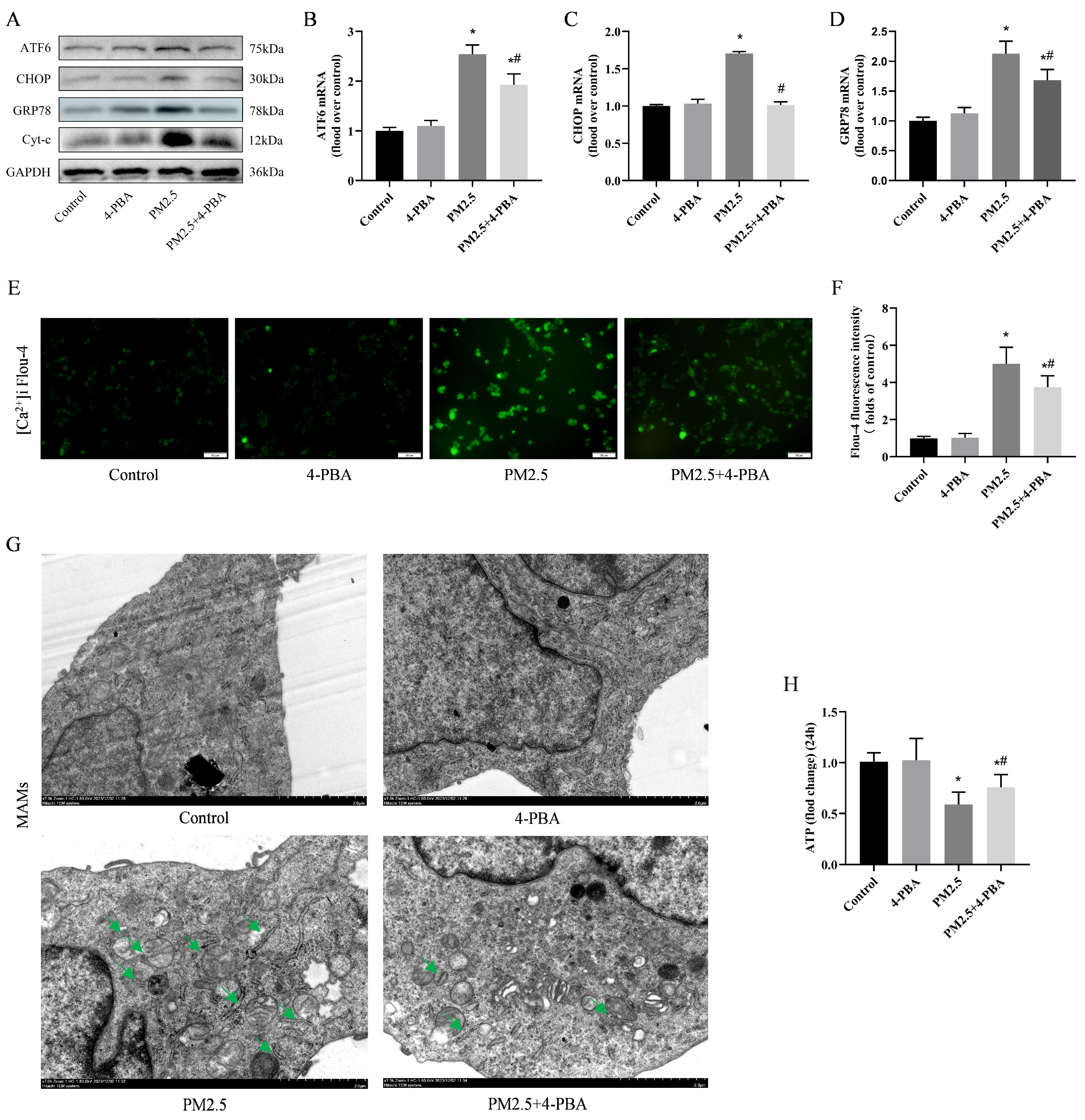
3.5. PM2.5-Induced Rat Alveolar Macrophage ER Stress and Mitochondrial Damage
3.6. Inhibition of ER Stress by 4-PBA Attenuated PM2.5-Induced [Ca2+]i Disorders and Alterations in MAMs
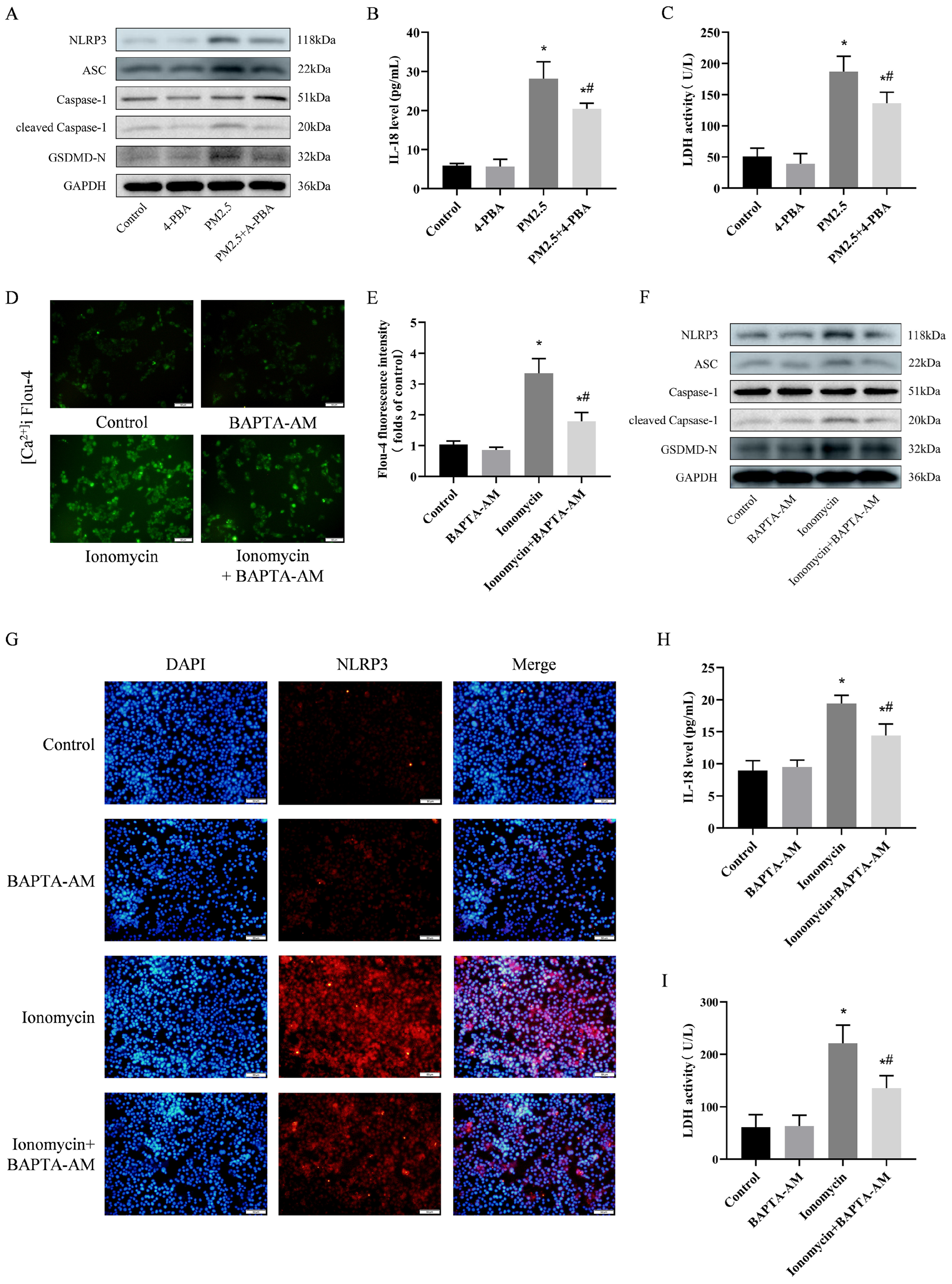
3.7. 4-PBA Inhibited PM2.5-Induced Cellular Inflammation and Pyroptosis
3.8. Preventing [Ca2+]i Disorder is an Important Pathway in Weakening NLRP3-Mediated Pyroptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Ma, T.; Chen, J.; Peng, X.; Zhang, Y.; Wang, Y.; Peng, J.; Shi, G.; Wei, Y.; Gao, J. Insights into PM2.5 pollution of four small and medium-sized cities in Chinese representative regions: Chemical compositions, sources and health risks. Sci. Total Environ. 2024, 918, 170620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, Y.; Su, Z.; Pu, W.; Niu, M.; Song, S.; Wei, L.; Ding, Y.; Xu, L.; Tian, M.; et al. Respiratory exposure to PM2.5 soluble extract disrupts mucosal barrier function and promotes the development of experimental asthma. Sci. Total Environ. 2020, 730, 139145. [Google Scholar] [CrossRef]
- Xin, H.; Gao, M.; Wang, X.; Qiu, T.; Guo, Y.; Zhang, L. Animal farms are hot spots for airborne antimicrobial resistance. Sci. Total Environ. 2022, 851, 158050. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, Z.; Hao, P.; Ji, S.; Gao, Y. Characteristics and health impacts of bioaerosols in animal barns: A comprehensive study. Ecotoxicol. Environ. Saf. 2024, 278, 116381. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Du, X.; Sun, Y.; Sun, K.; Zhang, X.; Wang, L.; Zhu, Y.; Basang, W.; Gao, Y. RGS2 attenuates alveolar macrophage damage by inhibiting the Gq/11-Ca2+ pathway during cowshed PM2.5 exposure, and aberrant RGS2 expression is associated with TLR2/4 activation. Toxicol. Appl. Pharmacol. 2024, 487, 116976. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, X.; Wu, D.; Wang, G.; Lan, H.; Zheng, X.; Li, S. IP3R1 is required for meiotic progression and embryonic development by regulating mitochondrial calcium and oxidative damage. Theriogenology 2024, 229, 147–157. [Google Scholar] [CrossRef]
- Loncke, J.; Kaasik, A.; Bezprozvanny, I.; Parys, J.B.; Kerkhofs, M.; Bultynck, G. Balancing ER-Mitochondrial Ca2+ Fluxes in Health and Disease. Trends Cell Biol. 2021, 31, 598–612. [Google Scholar] [CrossRef]
- Liu, H.; Lai, W.; Nie, H.; Shi, Y.; Zhu, L.; Yang, L.; Tian, L.; Li, K.; Bian, L.; Xi, Z.; et al. PM2.5 triggers autophagic degradation of Caveolin-1 via endoplasmic reticulum stress (ERS) to enhance the TGF-β1/Smad3 axis promoting pulmonary fibrosis. Environ. Int. 2023, 181, 108290. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, M. PM2.5 induces autophagy and apoptosis through endoplasmic reticulum stress in human endothelial cells. Sci. Total Environ. 2020, 710, 136397. [Google Scholar] [CrossRef]
- Xu, F.; Wang, L. Deciphering ER stress-unfolded protein response relationship by visualizing unfolded proteins in the ER. Cell Rep. 2024, 43, 114358. [Google Scholar] [CrossRef]
- Moonwiriyakit, A.; Dinsuwannakol, S.; Sontikun, J.; Timpratueang, K.; Muanprasat, C.; Khemawoot, P. Fine particulate matter PM2.5 and its constituent, hexavalent chromium induce acute cytotoxicity in human airway epithelial cells via inflammasome-mediated pyroptosis. Environ. Toxicol. Pharmacol. 2024, 107, 104416. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Tong, H.; Ren, C.; Chen, K.; Luo, S.; Wang, Q.; Guo, M.; Xu, Y.; Hu, M.; Fang, J.; et al. Inflammation unleashed: The role of pyroptosis in chronic liver diseases. Int. Immunopharmacol. 2024, 141, 113006. [Google Scholar] [CrossRef]
- Reddy, S.S.; Shruthi, K.; Joy, D.; Reddy, G.B. 4-PBA prevents diabetic muscle atrophy in rats by modulating ER stress response and ubiquitin-proteasome system. Chem. -Biol. Interact. 2019, 306, 70–77. [Google Scholar] [CrossRef]
- Zeng, M.; Sang, W.; Chen, S.; Chen, R.; Zhang, H.; Xue, F.; Li, Z.; Liu, Y.; Gong, Y.; Zhang, H.; et al. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol. Lett. 2017, 271, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Batibawa, J.W.; Du, Z.; Liang, S.; Duan, J.; Sun, Z. Acute exposure to PM2.5 triggers lung inflammatory response and apoptosis in rat. Ecotoxicol. Environ. Saf. 2021, 222, 112526. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pei, C.; Wang, X.; Wang, Y.; Huang, D.; Shi, S.; Shen, Z.; Li, S.; He, Y.; Wang, Z.; et al. Probiotics ameliorates pulmonary inflammation via modulating gut microbiota and rectifying Th17/Treg imbalance in a rat model of PM2.5 induced lung injury. Ecotoxicol. Environ. Saf. 2022, 244, 114060. [Google Scholar] [CrossRef]
- Wijagkanalan, W.; Kawakami, S.; Takenaga, M.; Igarashi, R.; Yamashita, F.; Hashida, M. Efficient targeting to alveolar macrophages by intratracheal administration of mannosylated liposomes in rats. J. Control. Release 2008, 125, 121–130. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. Actions of ionomycin, 4-BrA23187 and a novel electrogenic Ca2+ ionophore on mitochondria in intact cells. Cell Calcium 2003, 33, 101–112. [Google Scholar] [CrossRef]
- Esteves, G.N.N.; Ferraz, L.S.; Alvarez, M.M.P.; Costa, C.A.d.; Lopes, R.d.M.; Tersariol, I.L.d.S.; Rodrigues, T. BRAF and NRAS mutated melanoma: Different Ca2+ responses, Na+/Ca2+ exchanger expression, and sensitivity to inhibitors. Cell Calcium 2020, 90, 102241. [Google Scholar] [CrossRef]
- Porwisiak, P.; Werner, M.; Kryza, M.; ApSimon, H.; Woodward, H.; Mehlig, D.; Gawuc, L.; Szymankiewicz, K.; Sawiński, T. Application of ADMS-Urban for an area with a high contribution of residential heating emissions—Model verification and sensitivity study for PM2.5. Sci. Total Environ. 2024, 907, 168011. [Google Scholar] [CrossRef]
- Beaupied, B.L.; Martinez, H.; Martenies, S.; McConnel, C.S.; Pollack, I.B.; Giardina, D.; Fischer, E.V.; Jathar, S.; Duncan, C.G.; Magzamen, S. Cows as canaries: The effects of ambient air pollution exposure on milk production and somatic cell count in dairy cows. Environ. Res. 2022, 207, 112197. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, D.; Huang, L.; Guo, C.; Gao, X.; Xu, Z.; Yang, Z.; Chen, Y.; Li, M.; Yang, J. Global associations between long-term exposure to PM2.5 constituents and health: A systematic review and meta-analysis of cohort studies. J. Hazard. Mater. 2024, 474, 134715. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Kang, N.; Zhang, W.; Chen, B.; Xu, S.; Wu, L. The developmental toxicity of PM2.5 on the early stages of fetal lung with human lung bud tip progenitor organoids. Environ. Pollut. 2023, 330, 121764. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, W.; Yue, H.; Zhao, P.; Li, J. Effective-components combination alleviates PM2.5-induced inflammation by evoking macrophage autophagy in COPD. J. Ethnopharmacol. 2024, 321, 117537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, M.; Xiong, Y.; Zhang, L.; Xiong, A.; Wang, J.; He, X.; Li, G. Integrated analysis of ATAC-seq and RNA-seq unveils the role of ferroptosis in PM2.5-induced asthma exacerbation. Int. Immunopharmacol. 2023, 125, 111209. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Belmares, R.O.; Krais, A.M.; Esfahani, B.K.; Rosas-Pérez, I.; Mucs, D.; López-Marure, R.; Bergman, Å.; Alfaro-Moreno, E. Phthalate esters on urban airborne particles: Levels in PM10 and PM2.5 from Mexico City and theoretical assessment of lung exposure. Environ. Res. 2018, 161, 439–445. [Google Scholar] [CrossRef]
- Sun, J.; Yu, J.; Shen, Z.; Niu, X.; Wang, D.; Wang, X.; Xu, H.; Chuang, H.-C.; Cao, J.; Ho, K.-F. Oxidative stress–inducing effects of various urban PM2.5 road dust on human lung epithelial cells among 10 Chinese megacities. Ecotoxicol. Environ. Saf. 2021, 224, 112680. [Google Scholar] [CrossRef]
- Famiyeh, L.; Xu, H.; Chen, K.; Tang, Y.-T.; Ji, D.; Xiao, H.; Tong, L.; Jia, C.; Guo, Q.; He, J. Breathing in danger: Unveiling the link between human exposure to outdoor PM2.5-bound polycyclic aromatic hydrocarbons and lung cancer risk in an urban residential area of China. Sci. Total Environ. 2024, 907, 167762. [Google Scholar] [CrossRef]
- Sivakumar, B.; Kurian, G.A. Investigating the temporal link between PM2.5 exposure and acceleration of myocardial ischemia reperfusion injury: Emphasizing the hazardous presence of metals in inhaled air. Environ. Pollut. 2024, 355, 124113. [Google Scholar] [CrossRef]
- Zhao, S.; Li, H.; Yang, F.; Yang, Y.; Zeng, Y.; An, Z.; Li, J.; Wu, H.; Song, J.; Wu, W. Association of short-term PM2.5 exposure with airway innate immune response, microbiota and metabolism alterations in human airways. Environ. Pollut. 2024, 345, 123435. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Pan, W.-C.; Wu, C.-D.; Tsai, H.-J.; Yao, T.-C. Association between early postnatal exposure to PM2.5 and airway resistance among school-age children with asthma. J. Allergy Clin. Immunol. 2024, 153, AB190. [Google Scholar] [CrossRef]
- Min, Y.; Wei, X.; Yang, C.; Duan, Z.; Yang, J.; Ju, K.; Peng, X. Associations and attributable burdens in late-life exposure to PM2.5 and its major components and depressive symptoms in middle-aged and older adults: A nationwide cohort study. Ecotoxicol. Environ. Saf. 2024, 280, 116531. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cao, Z.; Jiao, X.; Bai, D.; Zhang, Y.; Hua, J.; Liu, W.; Teng, X. Pre-pregnancy exposure to fine particulate matter (PM2.5) increases reactive oxygen species production in oocytes and decrease litter size and weight in mice. Environ. Pollut. 2021, 268, 115858. [Google Scholar] [CrossRef]
- Aghaei-Zarch, S.M.; Nia, A.H.S.; Nouri, M.; Mousavinasab, F.; Najafi, S.; Bagheri-Mohammadi, S.; Aghaei-Zarch, F.; Toolabi, A.; Rasoulzadeh, H.; Ghanavi, J.; et al. The impact of particulate matters on apoptosis in various organs: Mechanistic and therapeutic perspectives. Biomed. Pharmacother. 2023, 165, 115054. [Google Scholar] [CrossRef]
- Chen, W.; Ge, P.; Lu, Z.; Liu, X.; Cao, M.; Yan, Z.; Chen, M. Acute exposure to seasonal PM2.5 induces toxicological responses in A549 cells cultured at the air-liquid interface mediated by oxidative stress and endoplasmic reticulum stress. Environ. Res. 2024, 248, 118283. [Google Scholar] [CrossRef]
- Pardo, M.; Xu, F.; Shemesh, M.; Qiu, X.; Barak, Y.; Zhu, T.; Rudich, Y. Nrf2 protects against diverse PM2.5 components-induced mitochondrial oxidative damage in lung cells. Sci. Total Environ. 2019, 669, 303–313. [Google Scholar] [CrossRef]
- Sun, K.; Sun, Y.; Jia, Y.; Duan, X.; Ma, Z.; Zhang, X.; Wang, L.; Zhu, Y.; Gao, Y.; Basang, W. MicroRNA miR-212-5p Regulates the MEK/ERK Signaling Pathway by Targeting A-Raf proto-oncogene serine/threonine-protein kinase (ARAF) to Regulate Cowshed PM2.5-Induced NR8383 Apoptosis. Toxics 2023, 11, 981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, S.; Fu, X.; Zhang, Q.; Wang, H. Emerging insights into the role of NLRP3 inflammasome and endoplasmic reticulum stress in renal diseases. Int. Immunopharmacol. 2024, 136, 112342. [Google Scholar] [CrossRef]
- Mou, Y.; Liao, W.; Liang, Y.; Li, Y.; Zhao, M.; Guo, Y.; Sun, Q.; Tang, J.; Wang, Z. Environmental pollutants induce NLRP3 inflammasome activation and pyroptosis: Roles and mechanisms in various diseases. Sci. Total Environ. 2023, 900, 165851. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Yang, J.; Cao, Z.; Hao, L.; Gu, Z. Exposure of nonylphenol promoted NLRP3 inflammasome and GSDMD-mediated pyroptosis in allergic rhinitis mice. Food Chem. Toxicol. 2024, 184, 114435. [Google Scholar] [CrossRef]
- Fan, X.; Lu, Q.; Jia, Q.; Li, L.; Cao, C.; Wu, Z.; Liao, M. Prevotella histicola ameliorates DSS-induced colitis by inhibiting IRE1α-JNK pathway of ER stress and NF-κB signaling. Int. Immunopharmacol. 2024, 135, 112285. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xie, M.; Liu, W.; Chen, S.; Ye, B.; Yao, J.; Xiao, Z.; Zhou, C.; Zheng, M. Inhibition of BTK improved APAP-induced liver injury via suppressing proinflammatory macrophages activation by restoring mitochondrion function. Int. Immunopharmacol. 2022, 110, 109036. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Chen, J.; Li, X.; Yang, R.; Cao, X.; Zhou, L.; Zhou, Y.; Ying, H.; Zhang, Q.; Sun, Y. Limonin ameliorates dextran sulfate sodium-induced chronic colitis in mice by inhibiting PERK-ATF4-CHOP pathway of ER stress and NF-κB signaling. Int. Immunopharmacol. 2021, 90, 107161. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-M.; Huang, T.-H.; Chi, M.-C.; Guo, S.-E.; Lee, C.-W.; Hwang, S.-L.; Shi, C.-S. N-acetylcysteine alleviates fine particulate matter (PM2.5)-induced lung injury by attenuation of ROS-mediated recruitment of neutrophils and Ly6Chigh monocytes and lung inflammation. Ecotoxicol. Environ. Saf. 2022, 239, 113632. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhang, Q.; Huang, G.; Huang, J.; Fu, X.; Liu, M.; Sun, Y.; Zhang, H. Vitamin B ameliorates PM2.5-induced kidney damage by reducing endoplasmic reticulum stress and oxidative stress in pregnant mice and HK-2. Toxicology 2023, 494, 153568. [Google Scholar] [CrossRef]
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca2+ Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39. [Google Scholar] [CrossRef]
- Sarg, N.H.; Zaher, D.M.; Abu Jayab, N.N.; Mostafa, S.H.; Ismail, H.H.; Omar, H.A. The interplay of p38 MAPK signaling and mitochondrial metabolism, a dynamic target in cancer and pathological contexts. Biochem. Pharmacol. 2024, 225, 116307. [Google Scholar] [CrossRef]
- Jia, H.; Zhang, T.; Liu, N.; Si, X.; Bai, J.; Yang, Y.; Chen, Z.; Wu, Z. 4-Phenylbutyric acid alleviates 3-acetyldeoxynivalenol-induced immune cells response by inhibiting endoplasmic reticulum stress in mouse spleen. Food Chem. Toxicol. 2022, 164, 113002. [Google Scholar] [CrossRef]
- Alharbi, A.F.; Parrington, J. Deciphering the Role of Endolysosomal Ca2+ Channels in Immunity. Front. Immunol. 2021, 12, 656965. [Google Scholar] [CrossRef]
- Wang, C.; Meng, X.; Meng, M.; Shi, M.; Sun, W.; Li, X.; Zhang, X.; Liu, R.; Fu, Y.; Song, L. Oxidative stress activates the TRPM2-Ca2+-NLRP3 axis to promote PM2.5-induced lung injury of mice. Biomed. Pharmacother. 2020, 130, 110481. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.P.S.; Ramos, J.M.O.; Gamaro, G.D.; Gioda, A.; Gioda, C.R.; Souza, I.C.C. Experimental rodent models exposed to fine particulate matter (PM2.5) highlighting the injuries in the central nervous system: A systematic review. Atmos. Pollut. Res. 2022, 13, 101407. [Google Scholar] [CrossRef]
- Li, J.; Tang, W.; Li, S.; He, C.; Dai, Y.; Feng, S.; Zeng, C.; Yang, T.; Meng, Q.; Meng, J.; et al. Ambient PM2.5 and its components associated with 10-year atherosclerotic cardiovascular disease risk in Chinese adults. Ecotoxicol. Environ. Saf. 2023, 263, 115371. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, D.; Liu, X.; Zhu, J.; Wang, F.; Li, B.; Li, L. Effects of PM2.5 exposure on reproductive system and its mechanisms. Chemosphere 2021, 264, 128436. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Z.; Du, X.; Sun, Y.; Jia, Y.; Liang, X.; Gao, Y. Attenuation of PM2.5-Induced Lung Injury by 4-Phenylbutyric Acid: Maintenance of [Ca2+]i Stability between Endoplasmic Reticulum and Mitochondria. Biomolecules 2024, 14, 1135. https://doi.org/10.3390/biom14091135
Ma Z, Du X, Sun Y, Jia Y, Liang X, Gao Y. Attenuation of PM2.5-Induced Lung Injury by 4-Phenylbutyric Acid: Maintenance of [Ca2+]i Stability between Endoplasmic Reticulum and Mitochondria. Biomolecules. 2024; 14(9):1135. https://doi.org/10.3390/biom14091135
Chicago/Turabian StyleMa, Zhenhua, Xiaohui Du, Yize Sun, Yunna Jia, Xiaojun Liang, and Yunhang Gao. 2024. "Attenuation of PM2.5-Induced Lung Injury by 4-Phenylbutyric Acid: Maintenance of [Ca2+]i Stability between Endoplasmic Reticulum and Mitochondria" Biomolecules 14, no. 9: 1135. https://doi.org/10.3390/biom14091135