
Feasibility of Ex Vivo Ligandomics

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Materials
2.2. Preparation of Single Retinal Cells
2.3. Immunopanning of Endothelial and Neuronal Cells
2.4. Purity Analysis of Immunopanned Cells
2.5. Ex Vivo Ligand Binding Assay
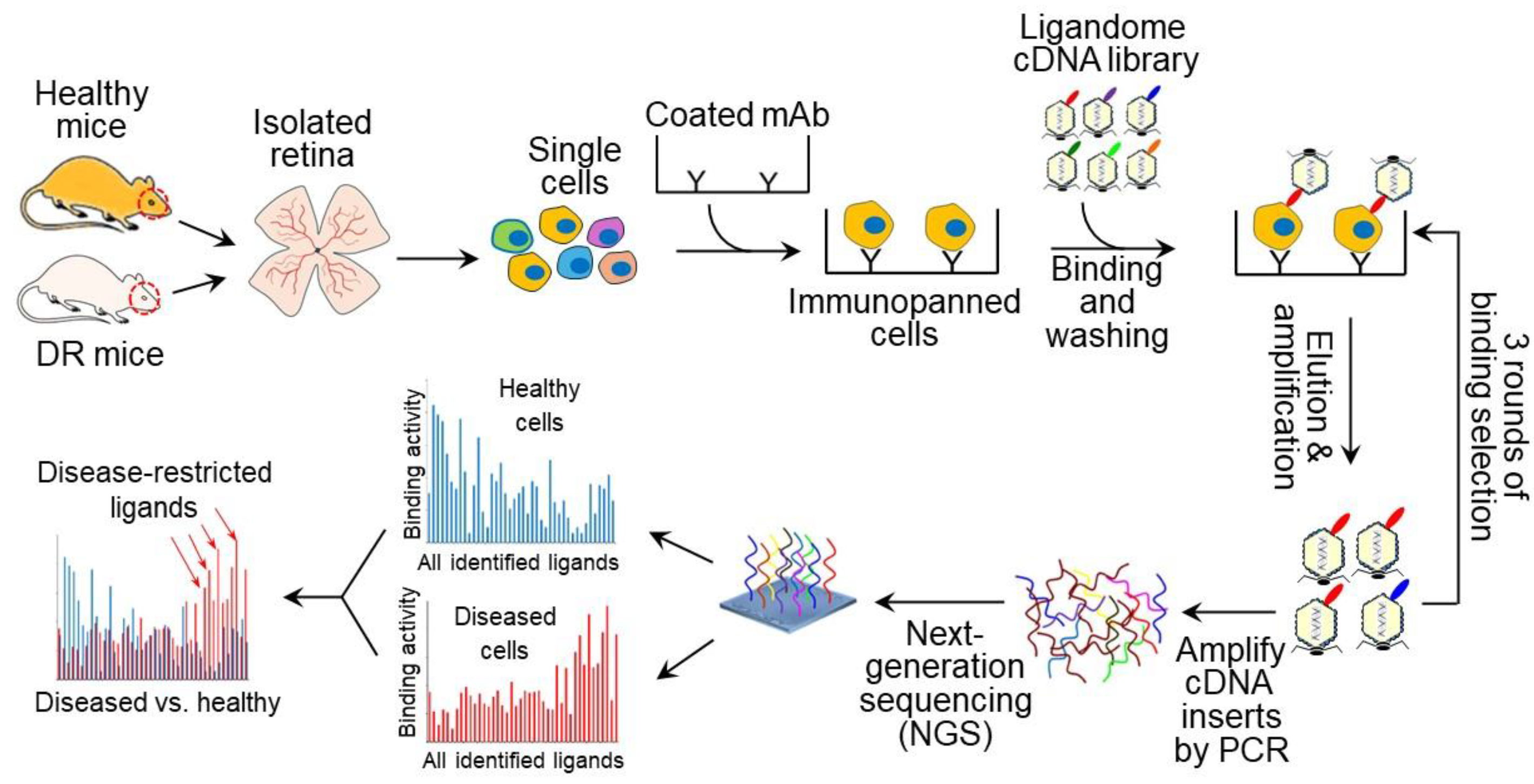
2.6. Ex Vivo Ligandomics
2.7. Statistical Analysis
3. Results
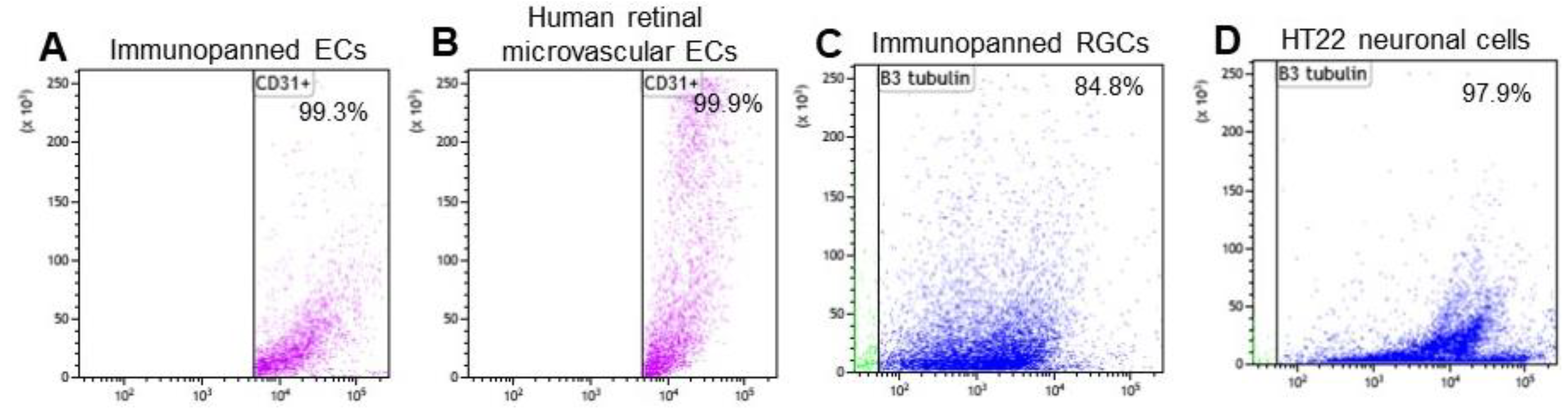
3.1. Cell Purity of Immunopanned ECs and RGCs
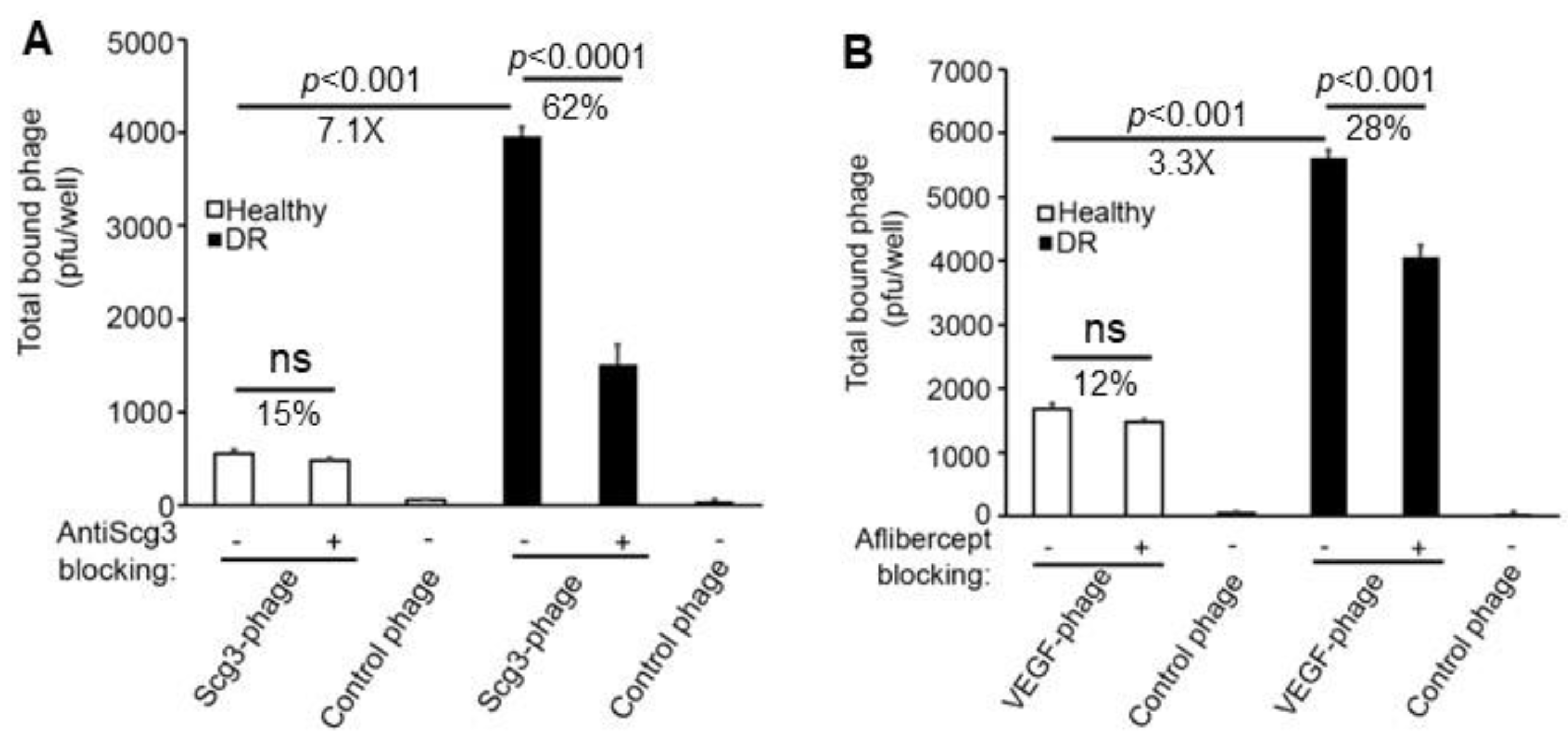
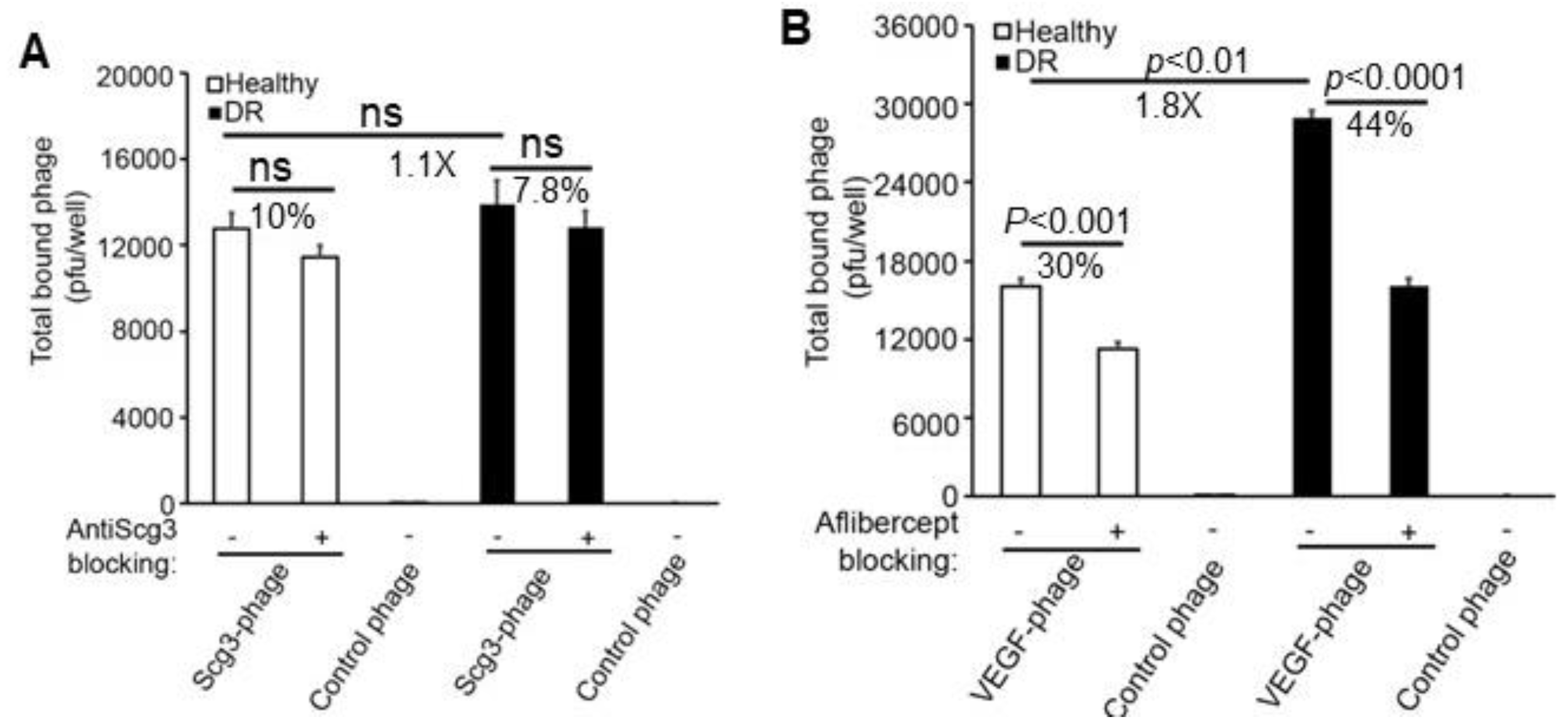
3.2. Binding of Scg3-Phage and VEGF-Phage to Immunopanned ECs
3.3. Binding of Scg3-Phage and VEGF-Phage to Immunopanned RGCs
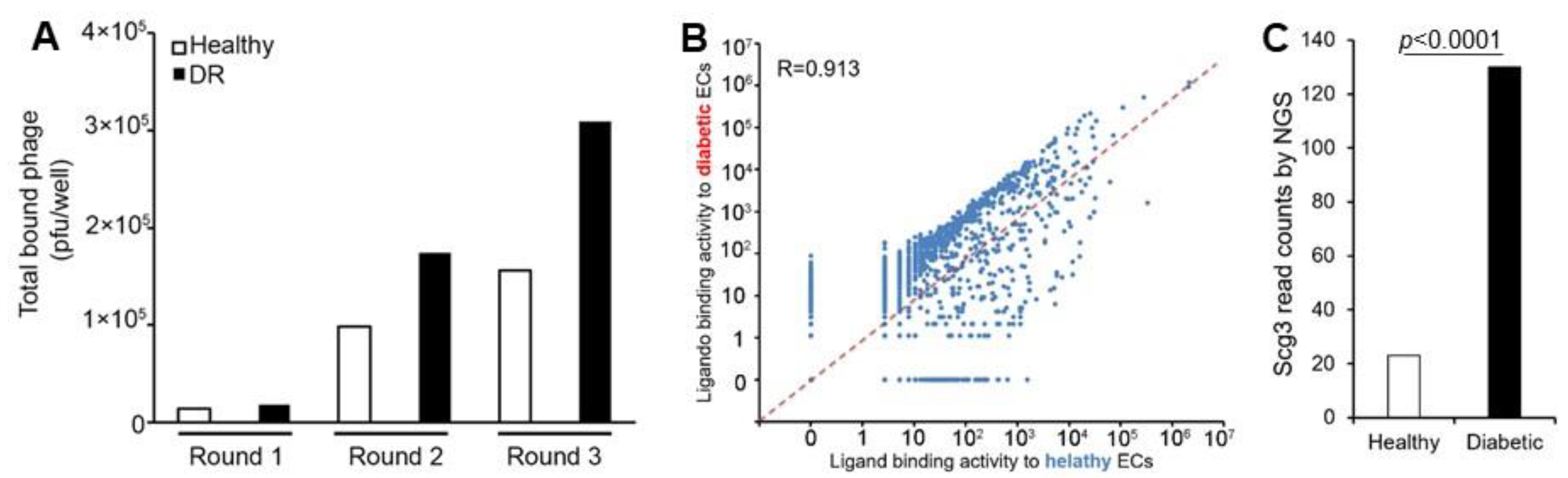
3.4. Ex Vivo Ligandomics Profiling Using Immunopanned ECs
3.5. Ex Vivo Ligandomics Profiling Using Immunopanned RGCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Combarnous, Y.; Nguyen, T.M.D. Cell Communications among Microorganisms, Plants, and Animals: Origin, Evolution, and Interplays. Int. J. Mol. Sci. 2020, 21, 8052. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Cochran, J.R. Targeting ligand-receptor interactions for development of cancer therapeutics. Curr. Opin. Chem. Biol. 2017, 38, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, I.; Hsu, S.Y.; Rauch, R.; Kowalski, H.W.; Hsueh, A.J.W. Signaling receptome: A genomic and evolutionary perspective of plasma membrane receptors involved in signal transduction. Sci. STKE 2003, 187, RE9. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, M.E.; Wang, W.; Chen, X.; Caberoy, N.B.; Guo, F.; Shen, C.; Ji, Y.; Tian, H.; Wang, H.; Chen, R.; et al. Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J. Exp. Med. 2017, 214, 1029–1047. [Google Scholar] [CrossRef]
- Waduge, P.; Tian, H.; Webster, K.A.; Li, W. Profiling disease-selective drug targets: From proteomics to ligandomics. Drug Discov. Today 2022, 28, 103430. [Google Scholar] [CrossRef]
- Ji, L.; Waduge, P.; Wan, W.; Tian, H.; Li, J.; Zhang, J.; Chen, R.; Li, W. Comparative ligandomics implicates secretogranin III as a disease-restricted angiogenic factor in laser-induced choroidal neovascularization. FEBS J. 2022, 289, 3521–3534. [Google Scholar] [CrossRef]
- Tang, F.; LeBlanc, M.E.; Wang, W.; Liang, D.; Chen, P.; Chou, T.-H.; Tian, H.; Li, W. Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis 2019, 22, 369–382. [Google Scholar] [CrossRef]
- Dai, C.; Waduge, P.; Ji, L.; Huang, C.; He, Y.; Tian, H.; Zuniga-Sanchez, E.; Bhatt, A.; Pang, I.-H.; Su, G.; et al. Secretogranin III stringently regulates pathological but not physiological angiogenesis in oxygen-induced retinopathy. Cell Mol. Life Sci. 2022, 79, 63. [Google Scholar] [CrossRef]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single dose streptozotocin-induced diabetes: Considerations for study design in islet transplantation models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef]
- Ji, L.; Waduge, P.; Wu, Y.; Huang, C.; Kaur, A.; Oliveira, P.; Tian, H.; Zhang, J.; Stout, J.T.; Weng, C.Y.; et al. Secretogranin III Selectively Promotes Vascular Leakage in the Deep Vascular Plexus of Diabetic Retinopathy. Int. J. Mol. Sci. 2023, 24, 10531. [Google Scholar] [CrossRef]
- Ji, L.; Waduge, P.; Hao, L.; Kaur, A.; Wan, W.; Wu, Y.; Tian, H.; Zhang, J.; Webster, K.A.; Li, W. Selectively targeting disease-restricted secretogranin III to alleviate choroidal neovascularization. FASEB J. 2022, 36, e22106. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Waduge, P.; Kaur, A.; Tian, H.; Weng, C.Y.; Stout, J.T.; Pang, I.-H.; Webster, K.A.; Li, W. Optimal Humanized Scg3-Neutralizing Antibodies for Anti-Angiogenic Therapy of Diabetic Retinopathy. Int. J. Mol. Sci. 2024, 25, 9507. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Shakya, A.; Wang, F.; Wu, W.D.; Li, W. Comparative Ligandomic Analysis of Human Lung Epithelial Cells Exposed to PM 2.5. Biomed. Environ. Sci. 2020, 33, 165–173. [Google Scholar] [PubMed]
- Privratsky, J.R.; Newman, P.J. PECAM-1: Regulator of endothelial junctional integrity. Cell Tissue Res. 2014, 355, 607–619. [Google Scholar] [CrossRef]
- Chintalapudi, S.R.; Djenderedjian, L.; Stiemke, A.B.; Steinle, J.J.; Jablonski, M.M.; Morales-Tirado, V.M. Isolation and Molecular Profiling of Primary Mouse Retinal Ganglion Cells: Comparison of Phenotypes from Healthy and Glaucomatous Retinas. Front. Aging Neurosci. 2016, 8, 93. [Google Scholar] [CrossRef]
- Waduge, P.; Kaur, A.; Li, W. A method for rapid and reliable quantification of VEGF-cell binding activity. Biochem. Biophys. Res. Commun. 2024, 727, 150321. [Google Scholar] [CrossRef]
- Sondell, M.; Sundler, F.; Kanje, M. Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur. J. Neurosci. 2000, 12, 4243–4254. [Google Scholar] [CrossRef]
- Li, W.; Pang, I.-H.; Pacheco, M.T.F.; Tian, H. Ligandomics: A paradigm shift in biological drug discovery. Drug Discov. Today 2018, 23, 636–643. [Google Scholar] [CrossRef]
- Rong, X.; Tian, H.; Yang, L.; Li, W. Function-first ligandomics for ocular vascular research and drug target discovery. Exp. Eye Res. 2019, 182, 57–64. [Google Scholar] [CrossRef]
- He, Y.; Tian, H.; Dai, C.; Wen, R.; Li, X.; Webster, K.A.; Li, W. Optimal Efficacy and Safety of Humanized Anti-Scg3 Antibody to Alleviate Oxygen-Induced Retinopathy. Int. J. Mol. Sci. 2021, 23, 350. [Google Scholar] [CrossRef]
- Richards, A.L.; Eckhardt, M.; Krogan, N.J. Mass spectrometry-based protein-protein interaction networks for the study of human diseases. Mol. Syst. Biol. 2021, 17, e8792. [Google Scholar] [CrossRef] [PubMed]
- Aizat, W.M.; Hassan, M. Proteomics in Systems Biology. Adv. Exp. Med. Biol. 2018, 1102, 31–49. [Google Scholar] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics. Nat. Protoc. 2024, 20, 180–219. [Google Scholar] [CrossRef]
- Su, C.J.; Murugan, A.; Linton, J.M.; Yeluri, A.; Bois, J.; Klumpe, H.; Langley, M.A.; Antebi, Y.E.; Elowitz, M.B. Ligand-receptor promiscuity enables cellular addressing. Cell Syst. 2022, 13, 408–425. [Google Scholar] [CrossRef]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef]
- Chen, G.; Gharib, T.G.; Huang, C.-C.; Taylor, J.M.G.; Misek, D.E.; Kardia, S.L.R.; Giordano, T.J.; Iannettoni, M.D.; Orringer, M.B.; Hanash, S.M.; et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell Proteom. 2002, 1, 304–313. [Google Scholar] [CrossRef]
- Kingsley, D.M.; Rinchik, E.M.; Russell, L.B.; Ottiger, H.P.; Sutcliffe, J.G.; Copeland, N.G.; Jenkins, N.A. Genetic ablation of a mouse gene expressed specifically in brain. EMBO J. 1990, 9, 395–399. [Google Scholar] [CrossRef]
- Morin, J.; Luu, T.M.; Superstein, R.; Ospina, L.H.; Lefebvre, F.; Simard, M.-N.; Shah, V.; Shah, P.S.; Kelly, E.N. Canadian Neonatal Network and the Canadian Neonatal Follow-Up Network Investigators. Neurodevelopmental Outcomes Following Bevacizumab Injections for Retinopathy of Prematurity. Pediatrics 2016, 137, e20153218. [Google Scholar] [CrossRef]
- Goldhardt, R.; Batawi, H.I.M.; Rosenblatt, M.; Lollett, I.V.; Park, J.J.; Galor, A. Effect of Anti-Vascular Endothelial Growth Factor Therapy on Corneal Nerves. Cornea 2019, 38, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Arima, M.; Akiyama, M.; Fujiwara, K.; Mori, Y.; Inoue, H.; Seki, E.; Nakama, T.; Tsukamoto, S.; Ochiai, M.; Ohga, S.; et al. Neurodevelopmental outcomes following intravitreal bevacizumab injection in Japanese preterm infants with type 1 retinopathy of prematurity. PLoS ONE 2020, 15, e0230678. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.; Ye, C.; Yang, Z.; Choi, B.; Chua, S.; Lowell, B.B. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 2011, 71, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Reichard, A.; Asosingh, K. Best Practices for Preparing a Single Cell Suspension from Solid Tissues for Flow Cytometry. Cytom. A 2019, 95, 219–226. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waduge, P.; Veettil, R.A.; Zhang, B.; Huang, C.; Tian, H.; Li, W. Feasibility of Ex Vivo Ligandomics. Biomolecules 2025, 15, 145. https://doi.org/10.3390/biom15010145
Waduge P, Veettil RA, Zhang B, Huang C, Tian H, Li W. Feasibility of Ex Vivo Ligandomics. Biomolecules. 2025; 15(1):145. https://doi.org/10.3390/biom15010145
Chicago/Turabian StyleWaduge, Prabuddha, Remya Ammassam Veettil, Bojun Zhang, Chengchi Huang, Hong Tian, and Wei Li. 2025. "Feasibility of Ex Vivo Ligandomics" Biomolecules 15, no. 1: 145. https://doi.org/10.3390/biom15010145
APA StyleWaduge, P., Veettil, R. A., Zhang, B., Huang, C., Tian, H., & Li, W. (2025). Feasibility of Ex Vivo Ligandomics. Biomolecules, 15(1), 145. https://doi.org/10.3390/biom15010145