
Application of Digital Polymerase Chain Reaction (dPCR) in Non-Invasive Prenatal Testing (NIPT)
 , and
, and
Abstract
:1. Introduction
2. The Role of cffDNA in NIPT: Biological Insights, Applications, and Technological Comparisons
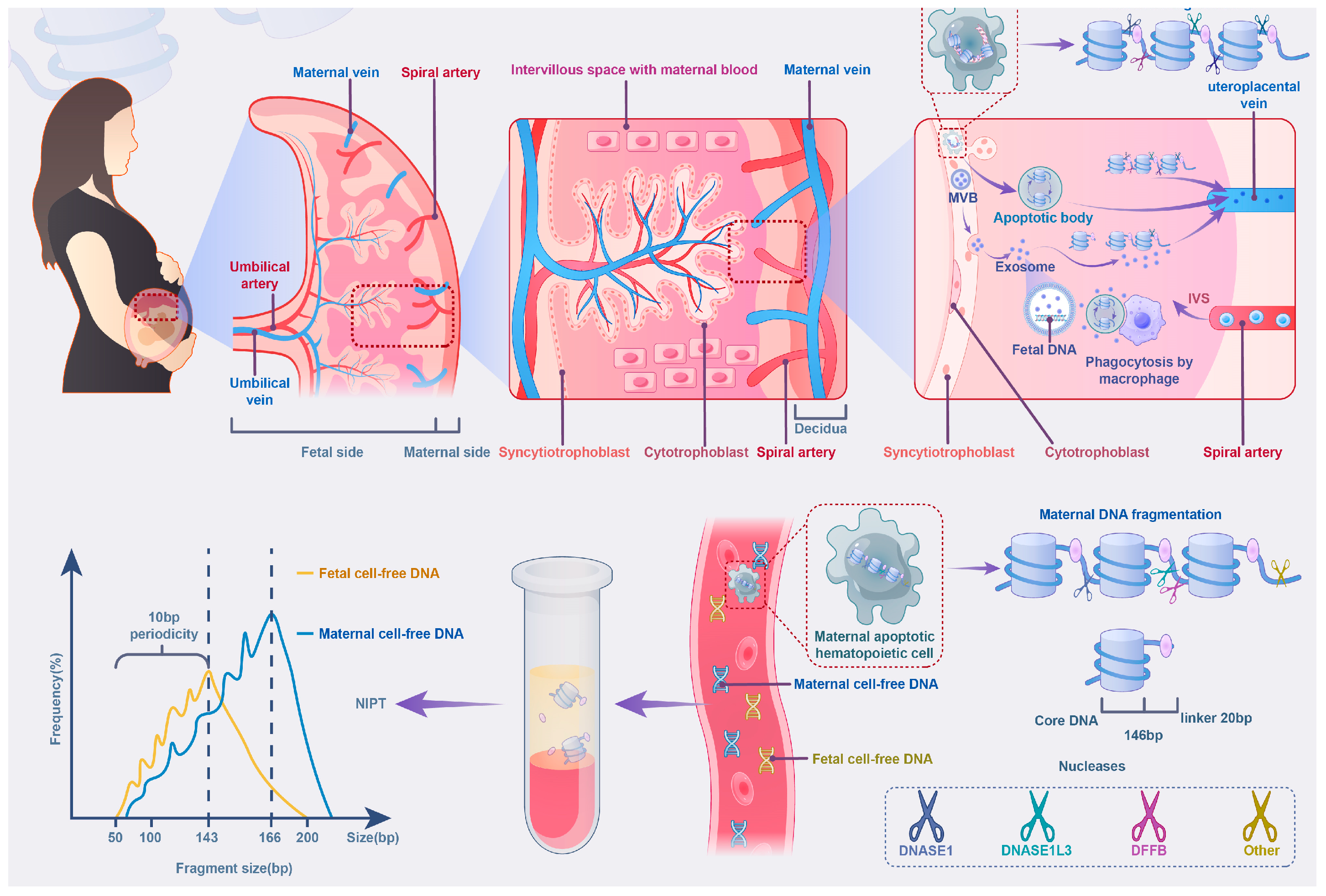
2.1. Fragmentation Characteristics and Molecular Stability of cffDNA
2.1.1. Preanalytical Factors Affecting cffDNA Quality and Detection
2.1.2. Comparison of NGS and dPCR in Utilizing cffDNA Characteristics
2.2. Placental Heterogeneity and the Regulation of cffDNA Release
2.2.1. Trophoblast Apoptosis and Oxygenation Impact on cffDNA Release
2.2.2. Role of Confined Placental Mosaicism (CPM)
2.2.3. Overcoming CPM Challenges with NGS and dPCR
2.3. Maternal Influences and Gestational Dynamics in cffDNA Release
NGS and dPCR: Addressing Maternal Interference Factors
3. From Maternal Serum Screening to NIPT with NGS and dPCR
3.1. Traditional Maternal Serum Screening
3.2. NIPT with NGS
3.3. NIPT with dPCR
3.4. Cost-Effectiveness Comparison of NGS and dPCR
3.5. Limitations in Detecting Certain Genetic Variations
4. The Working Principle of dPCR Technology for NIPT
4.1. Microfluidic Digital PCR (mdPCR)
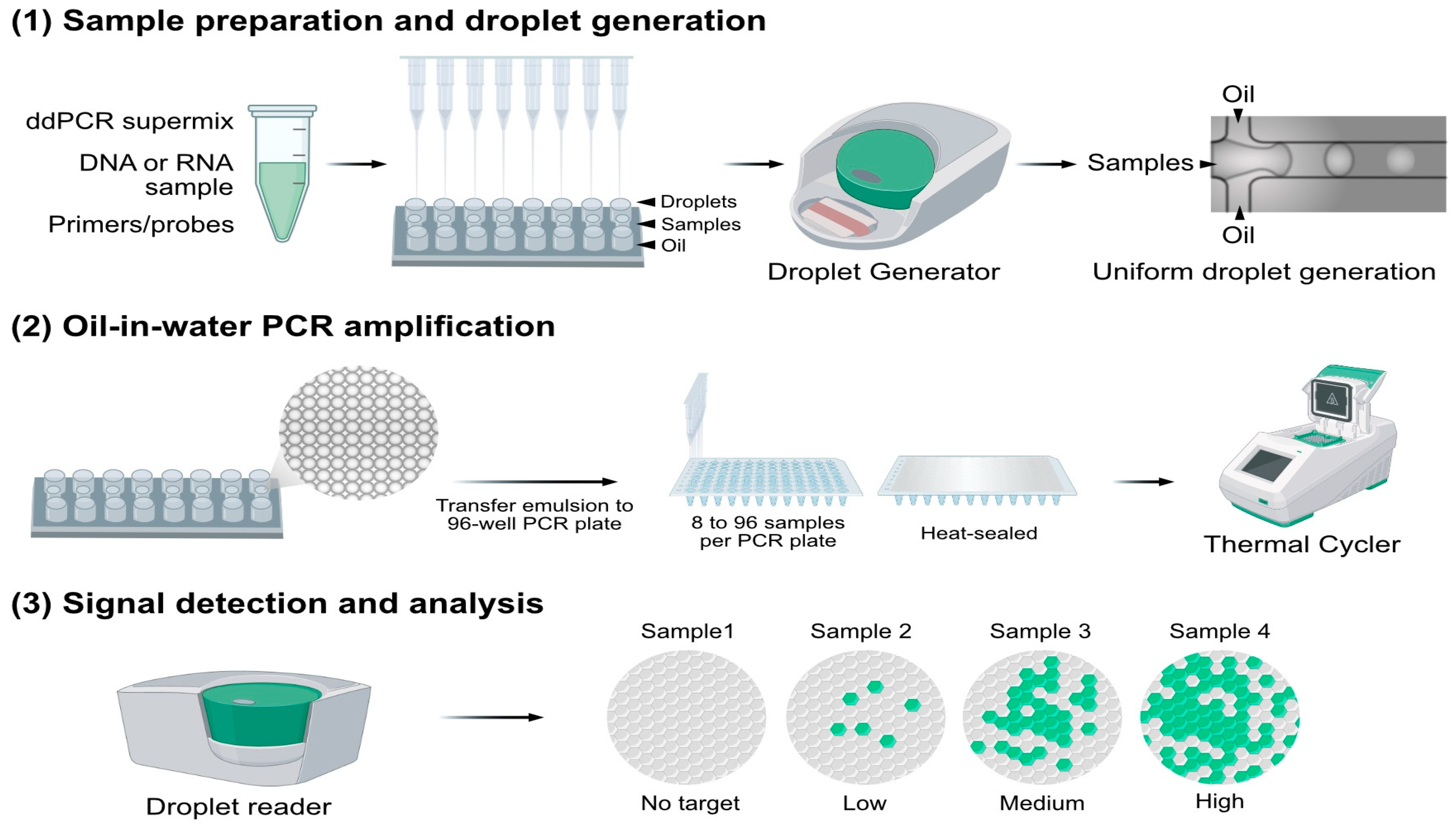
4.2. Droplet-Based Digital PCR (ddPCR)
4.3. Chip-Based Digital PCR (cdPCR)
5. The Role of dPCR in Prenatal Testing
6. Applications of dPCR in NIPT for Chromosomal Aneuploidy
6.1. Early Applications of dPCR in NIPT (2007–2015)
6.2. Advances in dPCR and the Development of Multiplex Detection (2015–2019)
6.3. Clinical Validation of dPCR in NIPT (2019–2021)
6.4. Technological Innovations and Future Directions of dPCR (2022–Present)
7. Clinical Applications of dPCR in NIPT for Chromosomal Microdeletions and Microduplications
8. Practical Applications of dPCR in NIPT for Monogenetic Disease
8.1. Fetal Blood System Monogenetic Diseases
8.1.1. Thalassemia
8.1.2. Sickle Cell Anemia
8.1.3. Hemophilia
8.2. Fetal Skeletal Muscle System Monogenetic Diseases
8.2.1. Achondroplasia
8.2.2. Spinal Muscular Atrophy (SMA)
8.3. Fetal Auditory System Monogenetic Disorders
Genetic Deafness
8.4. Fetal Respiratory and Digestive System Monogenetic Diseases
Cystic Fibrosis
8.5. Fetal Nervous System Monogenetic Diseases
Neurofibromatosis Type 1 (NF1)
9. Challenges in Detecting Maternally Inherited Mutations and the Role of dPCR
10. Prospects
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Akolekar, R.; Beta, J.; Picciarelli, G.; Ogilvie, C.; D’Antonio, F. Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: A systematic review and meta-analysis. Ultrasound Obs. Gynecol. 2015, 45, 16–26. [Google Scholar] [CrossRef]
- Lo, Y.M.; Hjelm, N.M.; Fidler, C.; Sargent, I.L.; Murphy, M.F.; Chamberlain, P.F.; Poon, P.M.; Redman, C.W.; Wainscoat, J.S. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N. Engl. J. Med. 1998, 339, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.M.; Benachi, A.; Gautier, E. New strategy for prenatal diagnosis of X-linked disorders. N. Engl. J. Med. 2002, 346, 1502. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.W.; Akolekar, R.; Zheng, Y.W.; Leung, T.Y.; Sun, H.; Chan, K.C.; Lun, F.M.; Go, A.T.; Lau, E.T.; To, W.W.; et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: Large scale validity study. BMJ. 2011, 342, c7401. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.K.; Chan, M.K.; Lo, P.S.; Chan, H.Y.; Chan, W.S.; Koo, T.Y.; Ng, H.Y.; Pooh, R.K. Clinical utility of noninvasive fetal trisomy (NIFTY) test--early experience. J. Matern. Fetal Neonatal Med. 2012, 25, 1856–1859. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Sayres, L.C.; Cho, M.K.; Cook-Deegan, R.; Chandrasekharan, S. Commercial landscape of noninvasive prenatal testing in the United States. Prenat. Diagn. 2013, 33, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, S.; Minear, M.A.; Hung, A.; Allyse, M. Noninvasive prenatal testing goes global. Sci. Transl. Med. 2014, 6, 231fs215. [Google Scholar] [CrossRef]
- Koumbaris, G.; Achilleos, A.; Nicolaou, M.; Loizides, C.; Tsangaras, K.; Kypri, E.; Mina, P.; Sismani, C.; Velissariou, V.; Christopoulou, G.; et al. Targeted capture enrichment followed by NGS: Development and validation of a single comprehensive NIPT for chromosomal aneuploidies, microdeletion syndromes and monogenic diseases. Mol. Cytogenet. 2019, 12, 48. [Google Scholar] [CrossRef]
- Xue, Y.; Zhao, G.; Qiao, L.; Lu, J.; Yu, B.; Wang, T. Sequencing Shorter cfDNA Fragments Decreases the False Negative Rate of Non-invasive Prenatal Testing. Front. Genet. 2020, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Sancha Dominguez, L.; Cotos Suarez, A.; Sanchez Ledesma, M.; Munoz Bellido, J.L. Present and Future Applications of Digital PCR in Infectious Diseases Diagnosis. Diagnostics 2024, 14, 931. [Google Scholar] [CrossRef] [PubMed]
- Olmedillas-Lopez, S.; Olivera-Salazar, R.; Garcia-Arranz, M.; Garcia-Olmo, D. Current and Emerging Applications of Droplet Digital PCR in Oncology: An Updated Review. Mol. Diagn. Ther. 2022, 26, 61–87. [Google Scholar] [CrossRef]
- Fan, H.C.; Quake, S.R. Detection of aneuploidy with digital polymerase chain reaction. Anal. Chem. 2007, 79, 7576–7579. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Costa, J.; Mafra, I. First nanoplate digital PCR method to trace allergenic foods: Improved sensitivity for the detection of sesame. Food Chem. 2024, 444, 138650. [Google Scholar] [CrossRef]
- Tan, L.L.; Loganathan, N.; Agarwalla, S.; Yang, C.; Yuan, W.; Zeng, J.; Wu, R.; Wang, W.; Duraiswamy, S. Current commercial dPCR platforms: Technology and market review. Crit. Rev. Biotechnol. 2023, 43, 433–464. [Google Scholar] [CrossRef]
- Wang, K.; Li, B.; Guo, Y.; Wu, Y.; Li, Y.; Wu, W. An integrated digital PCR system with high universality and low cost for nucleic acid detection. Front. Bioeng. Biotechnol. 2022, 10, 947895. [Google Scholar] [CrossRef]
- Tong, Y.; Shen, S.; Jiang, H.; Chen, Z. Application of Digital PCR in Detecting Human Diseases Associated Gene Mutation. Cell Physiol. Biochem. 2017, 43, 1718–1730. [Google Scholar] [CrossRef]
- Li, Y.Q.; Tan, G.J.; Zhou, Y.Q. Digital PCR and its applications in noninvasive prenatal testing. Brief. Funct. Genom. 2022, 21, 376–386. [Google Scholar] [CrossRef]
- Shekhawat, D.S.; Sharma, C.; Singh, K.; Singh, P.; Bhardwaj, A.; Patwa, P. Critical appraisal of droplet digital polymerase chain reaction application for noninvasive prenatal testing. Congenit. Anom. 2022, 62, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.C.; Lo, Y.M.D. Cell-Free DNA Fragmentomics in Liquid Biopsy. Diagnostics 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Stanley, K.E.; Jatsenko, T.; Tuveri, S.; Sudhakaran, D.; Lannoo, L.; Van Calsteren, K.; de Borre, M.; Van Parijs, I.; Van Coillie, L.; Van Den Bogaert, K.; et al. Cell type signatures in cell-free DNA fragmentation profiles reveal disease biology. Nat. Commun. 2024, 15, 2220. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Zhao, X.; Zhang, Z.; Xia, Z.; Yang, M.; Ma, L.; Zhao, Y.; Xu, G.; Du, S.; Wu, X.; et al. DNA methylation analysis explores the molecular basis of plasma cell-free DNA fragmentation. Nat. Commun. 2023, 14, 287. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Pan, M.; Shi, H.; Wang, L.; Bai, Y.; Ge, Q. Cell-Free DNA Fragmentomics: The Novel Promising Biomarker. Int. J. Mol. Sci. 2023, 24, 1503. [Google Scholar] [CrossRef]
- Han, D.S.C.; Ni, M.; Chan, R.W.Y.; Chan, V.W.H.; Lui, K.O.; Chiu, R.W.K.; Lo, Y.M.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef]
- Jiang, P.; Xie, T.; Ding, S.C.; Zhou, Z.; Cheng, S.H.; Chan, R.W.Y.; Lee, W.S.; Peng, W.; Wong, J.; Wong, V.W.S.; et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 2020, 30, 1144–1153. [Google Scholar] [CrossRef]
- Bai, K.; Lee, C.L.; Liu, X.; Li, J.; Cao, D.; Zhang, L.; Hu, D.; Li, H.; Hou, Y.; Xu, Y.; et al. Human placental exosomes induce maternal systemic immune tolerance by reprogramming circulating monocytes. J. Nanobiotechnol. 2022, 20, 86. [Google Scholar] [CrossRef]
- Yuen, N.; Lemaire, M.; Wilson, S.L. Cell-free placental DNA: What do we really know? PLoS Genet. 2024, 20, e1011484. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Holdenrieder, S. A pocket companion to cell-free DNA (cfDNA) preanalytics. Tumour Biol. 2024, 46, S297–S308. [Google Scholar] [CrossRef] [PubMed]
- Diaz, I.M.; Nocon, A.; Held, S.A.E.; Kobilay, M.; Skowasch, D.; Bronkhorst, A.J.; Ungerer, V.; Fredebohm, J.; Diehl, F.; Holdenrieder, S.; et al. Pre-Analytical Evaluation of Streck Cell-Free DNA Blood Collection Tubes for Liquid Profiling in Oncology. Diagnostics 2023, 13, 1288. [Google Scholar] [CrossRef] [PubMed]
- Krasic, J.; Abramovic, I.; Vrtaric, A.; Nikolac Gabaj, N.; Kralik-Oguic, S.; Katusic Bojanac, A.; Jezek, D.; Sincic, N. Impact of Preanalytical and Analytical Methods on Cell-Free DNA Diagnostics. Front. Cell Dev. Biol. 2021, 9, 686149. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Pan, M.; Zhou, Z.; Chen, C.; Xing, X.; Cheng, S.; Zhang, S.; Zheng, H.; Qian, K. The impact of preanalytical variables on the analysis of cell-free DNA from blood and urine samples. Front. Cell Dev. Biol. 2024, 12, 1385041. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, S.; Zhang, S.; He, Y.; Li, S.; Han, J.; Xu, M.; Deng, G. Performance of ImproGene Cell-Free DNA Tubes for Stabilization and Analysis of cfDNA in Blood Samples. Fetal Pediatr. Pathol. 2022, 41, 771–780. [Google Scholar] [CrossRef]
- Wreczycka, K.; Gosdschan, A.; Yusuf, D.; Gruning, B.; Assenov, Y.; Akalin, A. Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 2017, 261, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Chan, K.C.; Ding, C.; Gerovassili, A.; Yeung, S.W.; Chiu, R.W.; Leung, T.N.; Lau, T.K.; Chim, S.S.; Chung, G.T.; Nicolaides, K.H.; et al. Hypermethylated RASSF1A in maternal plasma: A universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin. Chem. 2006, 52, 2211–2218. [Google Scholar] [CrossRef]
- Lo, Y.M.; Zhang, J.; Leung, T.N.; Lau, T.K.; Chang, A.M.; Hjelm, N.M. Rapid clearance of fetal DNA from maternal plasma. Am. J. Hum. Genet. 1999, 64, 218–224. [Google Scholar] [CrossRef]
- Chiu, R.W.K.; Lo, Y.M.D. Cell-free fetal DNA coming in all sizes and shapes. Prenat. Diagn. 2021, 41, 1193–1201. [Google Scholar] [CrossRef]
- Schneider, L.; Tripathi, A. Sequence to size-based separation using microfluidic electrophoresis for targeted cell-free DNA analysis. Anal. Biochem. 2022, 649, 114691. [Google Scholar] [CrossRef] [PubMed]
- Breveglieri, G.; D’Aversa, E.; Finotti, A.; Borgatti, M. Non-Invasive Prenatal Testing Using Fetal DNA. Mol. Diagn. Ther. 2019, 23, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.C.H.; Vong, J.S.L.; Ji, L.; Poon, L.C.Y.; Jiang, P.; Lui, K.O.; Ni, Y.B.; To, K.F.; Cheng, Y.K.Y.; Chiu, R.W.K.; et al. Integrative single-cell and cell-free plasma RNA transcriptomics elucidates placental cellular dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, E7786–E7795. [Google Scholar] [CrossRef] [PubMed]
- Taglauer, E.S.; Wilkins-Haug, L.; Bianchi, D.W. Review: Cell-free fetal DNA in the maternal circulation as an indication of placental health and disease. Placenta. 2014, 35, S64–S68. [Google Scholar] [CrossRef] [PubMed]
- Farah, O.; Nguyen, C.; Tekkatte, C.; Parast, M.M. Trophoblast lineage-specific differentiation and associated alterations in preeclampsia and fetal growth restriction. Placenta 2020, 102, 4–9. [Google Scholar] [CrossRef]
- Bouvier, S.; Mousty, E.; Fortier, M.; Demattei, C.; Mercier, E.; Nouvellon, E.; Chea, M.; Grosjean, F.; Letouzey, V.; Gris, J.C. Placenta-mediated complications: Nucleosomes and free DNA concentrations differ depending on subtypes. J. Thromb. Haemost. 2020, 18, 3371–3380. [Google Scholar] [CrossRef]
- Neofytou, M. Predicting fetoplacental mosaicism during cfDNA-based NIPT. Curr. Opin. Obs. Gynecol. 2020, 32, 152–158. [Google Scholar] [CrossRef]
- Pittalis, M.C.; Dalpra, L.; Torricelli, F.; Rizzo, N.; Nocera, G.; Cariati, E.; Santarini, L.; Tibiletti, M.G.; Agosti, S.; Bovicelli, L.; et al. The predictive value of cytogenetic diagnosis after CVS based on 4860 cases with both direct and culture methods. Prenat Diagn. 1994, 14, 267–278. [Google Scholar] [CrossRef]
- Levy, B.; Hoffmann, E.R.; McCoy, R.C.; Grati, F.R. Chromosomal mosaicism: Origins and clinical implications in preimplantation and prenatal diagnosis. Prenat. Diagn. 2021, 41, 631–641. [Google Scholar] [CrossRef]
- Eggenhuizen, G.M.; Go, A.T.; Sauter, Z.; Hoffer, M.J.; Haak, M.C.; Geeven, G.; Diderich, K.E.; Joosten, M.; van den Born, M.; Srebniak, M.I. The role of confined placental mosaicism in fetal growth restriction: A retrospective cohort study. Prenat. Diagn. 2024, 44, 289–296. [Google Scholar] [CrossRef]
- Grati, F.R. Implications of fetoplacental mosaicism on cell-free DNA testing: A review of a common biological phenomenon. Ultrasound Obs. Gynecol. 2016, 48, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Qi, Q.; Zhou, X.; Jiang, Y.; Hao, N.; Liu, J. Factors associated with test failure in pregnant women undergoing cell-free DNA-based testing for fetal trisomy. J. Med. Screen. 2021, 28, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Zaki-Dizaji, M.; Shafiee, A.; Kohandel Gargari, O.; Fathi, H.; Heidary, Z. Maternal and Fetal Factors Affecting Cell-Free Fetal DNA (cffDNA) Fraction: A Systematic Review. J. Reprod. Infertil. 2023, 24, 219–231. [Google Scholar] [CrossRef]
- Hou, Y.; Yang, J.; Deng, F.; Wang, F.; Peng, H.; Guo, F.; Wang, D.; Yin, A. Association between cell-free DNA fetal fraction and pregnant character: A retrospective cohort study of 27,793 maternal plasmas. Sci. Rep. 2023, 13, 11420. [Google Scholar] [CrossRef]
- Stupak, A.; Kwasniewski, W.; Gozdzicka-Jozefiak, A.; Kwasniewska, A. The Influence of Maternal Obesity on Cell-Free Fetal DNA and Blood Pressure Regulation in Pregnancies with Hypertensive Disorders. Medicina 2021, 57, 962. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, L.; Zhang, J.; Song, Y. Efficacy and safety of low-dose aspirin combined with low-molecular-weight heparin in treatment of preeclampsia: A meta-analysis and systematic review. Arch. Med. Sci. 2022, 18, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.; Koelper, N.; Barberio, A.; Deagostino-Kelly, M.; Mennuti, M.; Sammel, M.D.; Dugoff, L. The association between anticoagulation therapy, maternal characteristics, and a failed cfDNA test due to a low fetal fraction. Prenat. Diagn. 2017, 37, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Gromminger, S.; Erkan, S.; Schock, U.; Stangier, K.; Bonnet, J.; Schloo, R.; Schubert, A.; Prott, E.C.; Knoll, U.; Stumm, M.; et al. The influence of low molecular weight heparin medication on plasma DNA in pregnant women. Prenat. Diagn. 2015, 35, 1155–1157. [Google Scholar] [CrossRef]
- Rink, B.D.; Stevens, B.K.; Norton, M.E. Incidental Detection of Maternal Malignancy by Fetal Cell-Free DNA Screening. Obs. Gynecol. 2022, 140, 121–131. [Google Scholar] [CrossRef]
- Schwaerzler, P. Controversies in Pregnancy Management after Prenatal Diagnosis of a Twin Pregnancy Discordant for Trisomy 21 Diagnosed by Cell-Free Fetal DNA Testing. SOJ Gynecol. Obstet. Women’s Health 2017, 3, 1–3. [Google Scholar] [CrossRef]
- Wang, E.; Batey, A.; Struble, C.; Musci, T.; Song, K.; Oliphant, A. Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat. Diagn. 2013, 33, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Hudecova, I.; Sahota, D.; Heung, M.M.; Jin, Y.; Lee, W.S.; Leung, T.Y.; Lo, Y.M.; Chiu, R.W. Maternal plasma fetal DNA fractions in pregnancies with low and high risks for fetal chromosomal aneuploidies. PLoS ONE 2014, 9, e88484. [Google Scholar] [CrossRef]
- Findley, T.O.; Parchem, J.G.; Ramdaney, A.; Morton, S.U. Challenges in the clinical understanding of genetic testing in birth defects and pediatric diseases. Transl. Pediatr. 2023, 12, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Wongkrajang, P.; Jittikoon, J.; Sangroongruangsri, S.; Talungchit, P.; Ruangvutilert, P.; Panchalee, T.; Chaikledkaew, U. Prenatal screening tests and prevalence of fetal aneuploidies in a tertiary hospital in Thailand. PLoS ONE 2023, 18, e0284829. [Google Scholar] [CrossRef]
- Praikaew, P.; Traisrisilp, K.; Wanapirak, C.; Sekararithi, R.; Tongsong, T. Ethnicity-Specific Normative Models of Quadruple Test as a Screening Test for Down Syndrome. Medicina 2021, 57, 651. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ning, W.; Shi, Y.; Chen, Y.; Zhang, W.; Li, L.; Wang, X. Maternal prenatal screening programs that predict trisomy 21, trisomy 18, and neural tube defects in offspring. PLoS ONE 2023, 18, e0281201. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; He, Y.; Xu, W.; Ma, Q.; He, Y.; Lei, W.; Chen, G.; He, Z.; Huang, J.; et al. Clinical performance of non-invasive prenatal served as a first-tier screening test for trisomy 21, 18, 13 and sex chromosome aneuploidy in a pilot city in China. Hum. Genom. 2020, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Zwiefelhofer, T.; Tim, R.C.; Dzakula, Z.; Kim, S.K.; Mazloom, A.R.; Zhu, Z.; Tynan, J.; Lu, T.; McLennan, G.; et al. High-throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS ONE 2013, 8, e57381. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.S.; Chen, Y.Y.; Ding, S.Y.; Zeng, L.; Shi, L.C.; Li, Y.J.; Zhang, J.J.; Fu, J.; Zhou, S.H.; He, J. Performance analysis of non-invasive prenatal testing for trisomy 13, 18, and 21: A large-scale retrospective study (2018–2021). Heliyon 2024, 10, e33437. [Google Scholar] [CrossRef]
- Li, C.; Xiong, M.; Zhan, Y.; Zhang, J.; Qiao, G.; Li, J.; Yang, H. Clinical Potential of Expanded Noninvasive Prenatal Testing for Detection of Aneuploidies and Microdeletion/Microduplication Syndromes. Mol. Diagn. Ther. 2023, 27, 769–779. [Google Scholar] [CrossRef]
- Zheng, J.; Lu, H.; Li, M.; Guan, Y.; Yang, F.; Xu, M.; Dong, J.; Zhang, Q.; An, N.; Zhou, Y. The Clinical Utility of Non-invasive Prenatal Testing for Pregnant Women With Different Diagnostic Indications. Front. Genet. 2020, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Parsaei, M.; Dashtkoohi, M.; Salmani, T.A.; Najafi, M.S.; Haddadi, M.; Ghaemi, M.; Hantoushzadeh, S. Potential efficacy of digital polymerase chain reaction for non-invasive prenatal screening of autosomal aneuploidies: A systematic review and meta-analysis. BMC Pregnancy Childbirth 2024, 24, 472. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Yang, Y.; Zhao, G.; Gu, Z.; Ren, H.; Hu, S.; Liu, N.; Jiao, W.; Li, J.; Kong, X. A dPCR-NIPT assay for detections of trisomies 21, 18 and 13 in a single-tube reaction-could it replace serum biochemical tests as a primary maternal plasma screening tool? J. Transl. Med. 2022, 20, 269. [Google Scholar] [CrossRef] [PubMed]
- Bayon, J.C.; Orruno, E.; Portillo, M.I.; Asua, J. The consequences of implementing non-invasive prenatal testing with cell-free foetal DNA for the detection of Down syndrome in the Spanish National Health Service: A cost-effectiveness analysis. Cost. Eff. Resour. Alloc. 2019, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mohammadi, T.; Sou, J.; Anis, A.H. Cost-effectiveness of prenatal screening and diagnostic strategies for Down syndrome: A microsimulation modeling analysis. PLoS ONE 2019, 14, e0225281. [Google Scholar] [CrossRef]
- Shang, W.; Wan, Y.; Chen, J.; Du, Y.; Huang, J. Introducing the non-invasive prenatal testing for detection of Down syndrome in China: A cost-effectiveness analysis. BMJ Open 2021, 11, e046582. [Google Scholar] [CrossRef]
- El Khattabi, L.A.; Rouillac-Le Sciellour, C.; Le Tessier, D.; Luscan, A.; Coustier, A.; Porcher, R.; Bhouri, R.; Nectoux, J.; Serazin, V.; Quibel, T.; et al. Could Digital PCR Be an Alternative as a Non-Invasive Prenatal Test for Trisomy 21: A Proof of Concept Study. PLoS ONE 2016, 11, e0155009. [Google Scholar] [CrossRef]
- Wei, X.; Lv, W.; Tan, H.; Liang, D.; Wu, L. Development and validation of a haplotype-free technique for non-invasive prenatal diagnosis of spinal muscular atrophy. J. Clin. Lab. Anal. 2020, 34, e23046. [Google Scholar] [CrossRef]
- Evans, M.I.; Sonek, J.D.; Hallahan, T.W.; Krantz, D.A. Cell-free fetal DNA screening in the USA: A cost analysis of screening strategies. Ultrasound Obs. Gynecol. 2015, 45, 74–83. [Google Scholar] [CrossRef]
- Hu, L.; Liang, F.; Cheng, D.; Zhang, Z.; Yu, G.; Zha, J.; Wang, Y.; Xia, Q.; Yuan, D.; Tan, Y.; et al. Location of Balanced Chromosome-Translocation Breakpoints by Long-Read Sequencing on the Oxford Nanopore Platform. Front. Genet. 2019, 10, 1313. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.A.; Nelson, E.T.; Koziura, M.; Cogan, J.D.; Hamid, R.; Phillips, J.A., 3rd. Lessons learned: Next-generation sequencing applied to undiagnosed genetic diseases. J. Clin. Investig. 2022, 132, e154942. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, S.E.; Hambuch, T.; Zook, J.M.; Bristow, S.L.; Hatchell, K.; Truty, R.; Kennemer, M.; Shirts, B.H.; Fellowes, A.; Chowdhury, S.; et al. One in seven pathogenic variants can be challenging to detect by NGS: An analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet. Med. 2021, 23, 1673–1680. [Google Scholar] [CrossRef]
- La Cognata, V.; Cavallaro, S. Detection of Structural Variants by NGS: Revealing Missing Alleles in Lysosomal Storage Diseases. Biomedicines 2022, 10, 1836. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, S.M.; Lawlor, J.M.J.; Handley, L.H.; Latner, D.R.; Bonnstetter, Z.T.; Finnila, C.R.; Thompson, M.L.; Boston, L.B.; Williams, M.; Rodriguez Nunez, I.; et al. Long-read genome sequencing and variant reanalysis increase diagnostic yield in neurodevelopmental disorders. Genome Res. 2024, 34, 1747–1762. [Google Scholar] [CrossRef]
- Newman, S.; Hermetz, K.E.; Weckselblatt, B.; Rudd, M.K. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet. 2015, 96, 208–220. [Google Scholar] [CrossRef]
- Quan, P.L.; Sauzade, M.; Brouzes, E. dPCR: A Technology Review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef]
- Tamura, T.; Imaizumi, T.; Shimojima Yamamoto, K.; Yamamoto, T. Genomic Copy Number Analysis Using Droplet Digital PCR: A Simple Method with EvaGreen Single-Color Fluorescent Design. Methods Mol. Biol. 2024, 2794, 293–304. [Google Scholar] [CrossRef]
- Wainman, L.M.; Sathyanarayana, S.H.; Lefferts, J.A. Applications of Digital Polymerase Chain Reaction (dPCR) in Molecular and Clinical Testing. J. Appl. Lab. Med. 2024, 9, 124–137. [Google Scholar] [CrossRef]
- Shestak, A.G.; Bukaeva, A.A.; Saber, S.; Zaklyazminskaya, E.V. Allelic Dropout Is a Common Phenomenon That Reduces the Diagnostic Yield of PCR-Based Sequencing of Targeted Gene Panels. Front. Genet. 2021, 12, 620337. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, M.; Usman, N.; Salnikova, E.; Sanakoeva, A.; Valiakhmetova, A.; Chervova, A.; Papusha, L.; Novichkova, G.; Druy, A. Methodological Challenges of Digital PCR Detection of the Histone H3 K27M Somatic Variant in Cerebrospinal Fluid. Pathol. Oncol. Res. 2022, 28, 1610024. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Chen, S.; Zheng, Y.; Zheng, X.; Lin, J.-M. Droplet-based digital PCR (ddPCR) and its applications. TrAC Trends Anal. Chem. 2023, 158, 116897. [Google Scholar] [CrossRef]
- Ren, Y.; Ji, J.; Zhang, H.; Cao, L.; Hu, J.; Xu, F.; Li, Z. A three-in-one microfluidic droplet digital PCR platform for absolute quantitative analysis of DNA. Lab. Chip. 2023, 23, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Howes, P.D.; deMello, A.J. Recent Advances in Droplet Microfluidics. Anal. Chem. 2020, 92, 132–149. [Google Scholar] [CrossRef]
- Jennings, L.J.; George, D.; Czech, J.; Yu, M.; Joseph, L. Detection and quantification of BCR-ABL1 fusion transcripts by droplet digital PCR. J. Mol. Diagn. 2014, 16, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Gou, T.; Hu, J.; Wu, W.; Ding, X.; Zhou, S.; Fang, W.; Mu, Y. Smartphone-based mobile digital PCR device for DNA quantitative analysis with high accuracy. Biosens. Bioelectron. 2018, 120, 144–152. [Google Scholar] [CrossRef]
- Madic, J.; Zocevic, A.; Senlis, V.; Fradet, E.; Andre, B.; Muller, S.; Dangla, R.; Droniou, M.E. Three-color crystal digital PCR. Biomol. Detect. Quantif. 2016, 10, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.; Lun, F.M.; Chan, K.C.; Tsui, N.B.; Chong, K.C.; Lau, T.K.; Leung, T.Y.; Zee, B.C.; Cantor, C.R.; Chiu, R.W. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc. Natl. Acad. Sci. USA 2007, 104, 13116–13121. [Google Scholar] [CrossRef]
- Lun, F.M.; Chiu, R.W.; Chan, K.C.; Leung, T.Y.; Lau, T.K.; Lo, Y.M. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin. Chem. 2008, 54, 1664–1672. [Google Scholar] [CrossRef]
- Xu, S.; Zou, B.; Xiang, Z.; Miao, M.; Song, Q.; Huang, H.; Wu, H.; Zhou, G. Non-invasive prenatal detection of trisomy 21 by quantifying segmental duplication in maternal plasma with digital PCR. Anal. Methods 2016, 8, 2138–2143. [Google Scholar] [CrossRef]
- Li, W.; Qian, L.; Qian, F.; Yu, X.; Yang, Z.; Wu, M. Application of droplet digital PCR for prenatal screening of Down syndrome. Clin. Exp. Obstet. Gynecol. 2018, 45, 231–236. [Google Scholar] [CrossRef]
- Lee, S.Y.; Shim, S.H.; Youn, J.-P.; Kim, S.J.; Kim, J.H.; Jung, S.A.; Choi, H.J.; Oh, M.J.; Lee, K.-R.; Cha, D.H.; et al. New application methods for chromosomal abnormalities screening test using digital PCR. BioChip J. 2015, 9, 339–352. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, S.J.; Han, S.H.; Park, J.S.; Choi, H.J.; Ahn, J.J.; Oh, M.J.; Shim, S.H.; Cha, D.H.; Hwang, S.Y. A new approach of digital PCR system for non-invasive prenatal screening of trisomy 21. Clin. Chim. Acta 2018, 476, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Chen, X.; Wang, F.; Wang, D.; Cao, Z.; Zhu, X.; Lu, C.; Yang, W.; Gao, N.; Gao, H.; et al. A multiplex droplet digital PCR assay for non-invasive prenatal testing of fetal aneuploidies. Analyst 2019, 144, 2239–2247. [Google Scholar] [CrossRef]
- Haidong, W.; Zhijie, Y.; Picchiassi, E.; Tarquini, F.; Coata, G.; You, W.; Youxiang, W.; Yu, C.; Di Renzo, G.C. Non-invasive prenatal testing of fetal aneuploidies using a new method based on digital droplet PCR and cell free fetal DNA. medRxiv, 2020. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Huang, Q.; Lin, X.; Wang, X.; Wang, Y.; Liu, Y.; He, Q.; Liu, Y.; Wang, T.; et al. Segmental duplication as potential biomarkers for non-invasive prenatal testing of aneuploidies. EBioMedicine 2021, 70, 103535. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Warczak, T.; Dzvova, N.; Abayan, R.; Gencoglu, M.; Riel, T.; Henriquez, A.; Markose, P.; Loomis, K.; Mikhaylichenko, O.; et al. P626: A novel, iteratively designed and highly multiplexed droplet digital PCR assay for non-invasive prenatal screening. Genet. Med. Open 2023, 1, 100682. [Google Scholar] [CrossRef]
- Lassakova, S.; Senkyrik, P.; Pazourkova, E.; Horinek, A.; Calda, P.; Brestak, M.; Svetnicova, K.; Neuzil, P.; Korabecna, M. Rapid non-invasive prenatal screening test for trisomy 21 based on digital droplet PCR. Sci. Rep. 2023, 13, 22948. [Google Scholar] [CrossRef]
- Hu, H.; Wang, L.; Wu, J.; Zhou, P.; Fu, J.; Sun, J.; Cai, W.; Liu, H.; Yang, Y. Noninvasive prenatal testing for chromosome aneuploidies and subchromosomal microdeletions/microduplications in a cohort of 8141 single pregnancies. Hum Genom. 2019, 13, 14. [Google Scholar] [CrossRef]
- Cillo, F.; Coppola, E.; Habetswallner, F.; Cecere, F.; Pignata, L.; Toriello, E.; De Rosa, A.; Grilli, L.; Ammendola, A.; Salerno, P.; et al. Understanding the Variability of 22q11.2 Deletion Syndrome: The Role of Epigenetic Factors. Genes. 2024, 15, 321. [Google Scholar] [CrossRef]
- Iordanescu, I.I.; Catana, A.; Cuzmici, Z.B.; Chelu, I.; Dragomir, C.; Militaru, M.; Severin, E.; Militaru, M.S. Microduplication and Microdeletion Syndromes Diagnosed Prenatally Using Single Nucleotide Polymorphism Array. J. Pers. Med. 2024, 14, 290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yue, F.; Zhang, X.; He, J.; Jiang, Y.; Liu, R.; Yu, Y. Prenatal detection of distal 1q21.1q21.2 microduplication with abnormal ultrasound findings: Two cases report and literature review. Medicine 2021, 100, e24227. [Google Scholar] [CrossRef] [PubMed]
- Zodanu, G.K.E.; Oszlanczi, M.; Havasi, K.; Kalapos, A.; Racz, G.; Katona, M.; Ujfalusi, A.; Nagy, O.; Szell, M.; Nagy, D. Systemic Screening for 22q11.2 Copy Number Variations in Hungarian Pediatric and Adult Patients With Congenital Heart Diseases Identified Rare Pathogenic Patterns in the Region. Front. Genet. 2021, 12, 635480. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Zhou, W.; Zhou, Y.; Zhou, L.; Wang, X.; Yu, B.; Zhang, B. Preliminary study of noninvasive prenatal screening for 22q11.2 deletion/duplication syndrome using multiplex dPCR assay. Orphanet J. Rare Dis. 2023, 18, 278. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.; Hartley, T.; Adam, S.; Bernier, F.; Chong, K.; Fernandez, B.A.; Friedman, J.M.; Geraghty, M.T.; Hume, S.; Knoppers, B.M.; et al. The clinical application of genome-wide sequencing for monogenic diseases in Canada: Position Statement of the Canadian College of Medical Geneticists. J. Med. Genet. 2015, 52, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.E.; Baker, J.M. Hemolytic Disease of the Fetus and Newborn: Historical and Current State. Clin. Lab. Med. 2021, 41, 133–151. [Google Scholar] [CrossRef]
- Patel, P.; Risler, Z.; Hobbib, G. Life threatening hemorrhage in hemophilia A. Vis. J. Emerg. Med. 2023, 31, 101660. [Google Scholar] [CrossRef]
- Vlachodimitropoulou, E.; Mogharbel, H.; Kuo, K.H.M.; Hwang, M.; Ward, R.; Shehata, N.; Malinowski, A.K. Pregnancy outcomes and iron status in beta-thalassemia major and intermedia: A systematic review and meta-analysis. Blood Adv. 2024, 8, 746–757. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C. Updated Worldwide Epidemiology of Inherited Erythrocyte Disorders. Acta Haematol. 2020, 143, 196–203. [Google Scholar] [CrossRef]
- Jaing, T.H.; Chang, T.Y.; Chen, S.H.; Lin, C.W.; Wen, Y.C.; Chiu, C.C. Molecular genetics of beta-thalassemia: A narrative review. Medicine 2021, 100, e27522. [Google Scholar] [CrossRef]
- Musallam, K.M.; Lombard, L.; Kistler, K.D.; Arregui, M.; Gilroy, K.S.; Chamberlain, C.; Zagadailov, E.; Ruiz, K.; Taher, A.T. Epidemiology of clinically significant forms of alpha- and beta-thalassemia: A global map of evidence and gaps. Am. J. Hematol. 2023, 98, 1436–1451. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Traisrisilp, K.; Sirichotiyakul, S.; Phusua, A.; Sanguansermsri, T.; Tongsong, T. Noninvasive Prenatal Diagnosis of Beta-Thalassemia Disease by Using Digital PCR Analysis of Cell-Free Fetal DNA in Maternal Plasma. Fetal Diagn. Ther. 2022, 49, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Sawakwongpra, K.; Tangmansakulchai, K.; Ngonsawan, W.; Promwan, S.; Chanchamroen, S.; Quangkananurug, W.; Sriswasdi, S.; Jantarasaengaram, S.; Ponnikorn, S. Droplet-based digital PCR for non-invasive prenatal genetic diagnosis of alpha and beta-thalassemia. Biomed. Rep. 2021, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Suwannakhon, N.; Hemvuthiphan, J.; Pangeson, T.; Mahingsa, K.; Pingyod, A.; Bumrungpakdee, W.; Sanguansermsri, T. Non-invasive prenatal screening & diagnosis of beta-thalassaemia in an affected foetus. Indian. J. Med. Res. 2023, 157, 447–452. [Google Scholar] [CrossRef]
- D’Aversa, E.; Breveglieri, G.; Boutou, E.; Balassopoulou, A.; Voskaridou, E.; Pellegatti, P.; Guerra, G.; Scapoli, C.; Gambari, R.; Borgatti, M. Droplet Digital PCR for Non-Invasive Prenatal Detection of Fetal Single-Gene Point Mutations in Maternal Plasma. Int. J. Mol. Sci. 2022, 23, 2819. [Google Scholar] [CrossRef]
- Barrett, A.N.; McDonnell, T.C.; Chan, K.C.; Chitty, L.S. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin. Chem. 2012, 58, 1026–1032. [Google Scholar] [CrossRef]
- Tsui, N.B.; Kadir, R.A.; Chan, K.C.; Chi, C.; Mellars, G.; Tuddenham, E.G.; Leung, T.Y.; Lau, T.K.; Chiu, R.W.; Lo, Y.M. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA. Blood 2011, 117, 3684–3691. [Google Scholar] [CrossRef]
- Hudecova, I.; Jiang, P.; Davies, J.; Lo, Y.M.D.; Kadir, R.A.; Chiu, R.W.K. Noninvasive detection of F8 int22h-related inversions and sequence variants in maternal plasma of hemophilia carriers. Blood 2017, 130, 340–347. [Google Scholar] [CrossRef]
- Orhant, L.; Anselem, O.; Fradin, M.; Becker, P.H.; Beugnet, C.; Deburgrave, N.; Tafuri, G.; Letourneur, F.; Goffinet, F.; Allach El Khattabi, L.; et al. Droplet digital PCR combined with minisequencing, a new approach to analyze fetal DNA from maternal blood: Application to the non-invasive prenatal diagnosis of achondroplasia. Prenat. Diagn. 2016, 36, 397–406. [Google Scholar] [CrossRef]
- Pacault, M.; Verebi, C.; Lopez, M.; Vaucouleur, N.; Orhant, L.; Deburgrave, N.; Leturcq, F.; Vidaud, D.; Girodon, E.; Bienvenu, T.; et al. Non-invasive prenatal diagnosis of single gene disorders by paternal mutation exclusion: 3 years of clinical experience. BJOG 2022, 129, 1879–1886. [Google Scholar] [CrossRef]
- Chang, M.Y.; Ahn, S.; Kim, M.Y.; Han, J.H.; Park, H.R.; Seo, H.K.; Yoon, J.; Lee, S.; Oh, D.Y.; Kang, C.; et al. One-step noninvasive prenatal testing (NIPT) for autosomal recessive homozygous point mutations using digital PCR. Sci. Rep. 2018, 8, 2877. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.; Pacault, M.; El Khattabi, L.A.; Vaucouleur, N.; Orhant, L.; Bienvenu, T.; Girodon, E.; Vidaud, D.; Leturcq, F.; Costa, C.; et al. Non-invasive prenatal diagnosis of paternally inherited disorders from maternal plasma: Detection of NF1 and CFTR mutations using droplet digital PCR. Clin. Chem. Lab. Med. 2018, 56, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Debrand, E.; Lykoudi, A.; Bradshaw, E.; Allen, S.K. A Non-Invasive Droplet Digital PCR (ddPCR) Assay to Detect Paternal CFTR Mutations in the Cell-Free Fetal DNA (cffDNA) of Three Pregnancies at Risk of Cystic Fibrosis via Compound Heterozygosity. PLoS ONE 2015, 10, e0142729. [Google Scholar] [CrossRef] [PubMed]
- Arishi, W.A.; Alhadrami, H.A.; Zourob, M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines 2021, 12, 519. [Google Scholar] [CrossRef]
- Serjeant, G.R. The geography of sickle cell disease: Opportunities for understanding its diversity. Ann. Saudi Med. 1994, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Kunicki, T.; Lillicrap, D. Genetic sequence analysis of inherited bleeding diseases. Blood 2013, 122, 3423–3431. [Google Scholar] [CrossRef]
- Zimmerman, B.; Valentino, L.A. Hemophilia: In review. Pediatr. Rev. 2013, 34, 289–294; quiz 295. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cao, J.; Shi, X.; Li, Y.; Qiao, F.; Wu, Y. Genetic testing and diagnostic strategies of fetal skeletal dysplasia: A preliminary study in Wuhan, China. Orphanet J. Rare Dis. 2023, 18, 336. [Google Scholar] [CrossRef]
- Tripathi, N.; Singh, A.; Agarwal, P. Worsening anasarca on a child with severe steroid-dependent nephrotic syndrome without proteinuria: Answers. Pediatr. Nephrol. 2021, 36, 4019–4020. [Google Scholar] [CrossRef]
- Savarirayan, R. Advances in the management of achondroplasia. Nat. Rev. Endocrinol. 2024, 20, 443–444. [Google Scholar] [CrossRef]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Yan, Y.; Guo, N.; Wang, F.; Wang, S.; Zhu, L.; Wang, Y.; Ma, Y.; Guo, Y. Single-Tube Multiplex Digital Polymerase Chain Reaction Assay for Molecular Diagnosis and Prediction of Severity of Spinal Muscular Atrophy. Anal. Chem. 2022, 94, 3517–3525. [Google Scholar] [CrossRef]
- Wener, E.R.; McLennan, J.D.; Papsin, B.C.; Cushing, S.L.; Stavropoulos, D.J.; Mendoza-Londono, R.; Quercia, N.; Gordon, K.A. Variants in Genes Associated with Hearing Loss in Children: Prevalence in a Large Canadian Cohort. Laryngoscope 2024, 134, 3832–3838. [Google Scholar] [CrossRef] [PubMed]
- Fromme, M.; Schneider, C.V.; Trautwein, C.; Brunetti-Pierri, N.; Strnad, P. Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J. Hepatol. 2022, 76, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Zani, A.; Chung, W.K.; Deprest, J.; Harting, M.T.; Jancelewicz, T.; Kunisaki, S.M.; Patel, N.; Antounians, L.; Puligandla, P.S.; Keijzer, R. Congenital diaphragmatic hernia. Nat. Rev. Dis. Primers 2022, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, B.A.; Turcios, N.L. Pulmonary Manifestations of Genetic Disorders in Children. Pediatr. Clin. N. Am. 2021, 68, 1–24. [Google Scholar] [CrossRef]
- Lopez-Valdez, J.A.; Aguilar-Alonso, L.A.; Gandara-Quezada, V.; Ruiz-Rico, G.E.; Avila-Soledad, J.M.; Reyes, A.A.; Pedroza-Jimenez, F.D. Cystic fibrosis: Current concepts. Bol. Med. Hosp. Infant. Mex. 2021, 78, 584–596. [Google Scholar] [CrossRef]
- Vgontzas, A.; Renthal, W. Introduction to Neurogenetics. Am. J. Med. 2019, 132, 142–152. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Ye, J.; Chen, C.; Yuan, Y.; Han, L.; Wang, Y.; Qiu, W.; Zhang, H.; Asan; Gu, X. Haplotype-based Noninvasive Prenatal Diagnosis of Hyperphenylalaninemia through Targeted Sequencing of Maternal Plasma. Sci. Rep. 2018, 8, 161. [Google Scholar] [CrossRef]
- Han, D.S.C.; Lo, Y.M.D. Non-invasive prenatal testing of monogenic fetal characteristics by maternal plasmaDNAanalysis. ISBT Sci. Ser. 2015, 10, 197–205. [Google Scholar] [CrossRef]
- Tsui, W.H.A.; Ding, S.C.; Jiang, P.; Lo, Y.M.D. Artificial intelligence and machine learning in cell-free-DNA-based diagnostics. Genome Res. 2025, 35, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, S.M.; Ahn, J.M.; Lee, T.R.; Kim, W.; Cho, E.H.; Ki, C.S. Development and performance evaluation of an artificial intelligence algorithm using cell-free DNA fragment distance for non-invasive prenatal testing (aiD-NIPT). Front. Genet. 2022, 13, 999587. [Google Scholar] [CrossRef] [PubMed]
- Liscovitch-Brauer, N.; Mesika, R.; Rabinowitz, T.; Volkov, H.; Grad, M.; Matar, R.T.; Basel-Salmon, L.; Tadmor, O.; Beker, A.; Shomron, N. Machine learning-enhanced noninvasive prenatal testing of monogenic disorders. Prenat. Diagn. 2024, 44, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Qiao, Y.; Tu, J. Microfluidic Technologies for cfDNA Isolation and Analysis. Micromachines 2019, 10, 672. [Google Scholar] [CrossRef]
- Guo, K.; Song, Z.; Zhou, J.; Shen, B.; Yan, B.; Gu, Z.; Wang, H. An artificial intelligence-assisted digital microfluidic system for multistate droplet control. Microsyst. Nanoeng. 2024, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Bellair, M.; Amaral, E.; Ouren, M.; Roark, C.; Kim, J.; O’Connor, A.; Soriano, A.; Schindler, M.L.; Wapner, R.J.; Stone, J.L.; et al. Noninvasive single-cell-based prenatal genetic testing: A proof of concept clinical study. Prenat. Diagn. 2024, 44, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Liu, X.; Maiga, M.; Cao, W.; Mu, Y.; Yan, Q.; Zhu, Q. Digital PCR for Single-Cell Analysis. Biosensors 2024, 14, 64. [Google Scholar] [CrossRef]
- Wang, C.; Qiu, J.; Liu, M.; Wang, Y.; Yu, Y.; Liu, H.; Zhang, Y.; Han, L. Microfluidic Biochips for Single-Cell Isolation and Single-Cell Analysis of Multiomics and Exosomes. Adv. Sci. 2024, 11, e2401263. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Author (Year) | Focus of Study | Method Used | Sample Information | Key Findings/Implications | Diagnostic Accuracy | Major Limitations |
|---|---|---|---|---|---|---|
| Lo YM et al. (2007) [99] | T21 | ddPCR | Normal = 9, T21 plasma = 4; normal = 2, T21 placenta = 2; GA: NA | Identified chromosomal imbalances through SNPs in the PLAC4 gene and chromosome dosage | The RNA and DNA from both plasma and placental samples were all correctly classified | Requires high FF (≥25%); applicable only to SNP-based detection; labor-intensive process |
| Fan HC et al. (2007) [16] | T21 | mdPCR | Human genomic DNA from normal and T21 cell lines; GA: NA | Detected T21 by amplifying and quantifying single DNA molecules in mixed samples | T21 can be distinguished with maternal contamination or fetal mosaicism | High DNA input (≥103 copies required); low sensitivity (≥10% fetal DNA needed) |
| El Khattabi LA et al. (2016) [77] | T21 | ddPCR | Normal = 192, T21 = 21; GA: 9–37 weeks | Detected T21 with 5% fetal DNA content | Sensitivity = 94%; specificity = 98% | Exclusion of 9 low-quality samples (4 T21) may overestimate sensitivity; no FF estimation (impacts reliability) |
| Xu S et al. (2016) [101] | T21 | cdPCR | Normal = 12, T21 = 3; GA: NA | Quantified small increases in Chr21 with 10% fetal DNA content | Accuracy = 100% | Inferred FF: reducing reliability; SD-based detection prone to amplification bias |
| Li W et al. (2018) [102] | T21 | ddPCR | Normal = 78, T21 = 28; GA: second trimester | ddPCR with HLCS gene and SNP rs6636 differentiates euploid from T21, showing significant ratio differences | Euploid: accuracy = 100%; T21: false negative = 2 cases | Only second trimester samples; unverified BstUI digestion; ethnic variability in SNP rs663 |
| Lee SY et al. (2015) [103] | T21, T18 | cdPCR | Low-risk samples = 28, T21 = 10, T18 = 5; GA: 10–35 weeks | cdPCR shows no cross-reactivity in T21 and T18 detection, enabling detection of chromosomal abnormalities at a 1.38% fragment ratio | T21: accuracy = 90%; T18: accuracy = 100% | Cut-off: empirical threshold, not statistically validated; FF: qPCR-based, lacks fetal specificity |
| Lee SY et al. (2018) [104] | T21 | ddPCR | Normal = 827, T21 = 50; GA: 10–22+3 weeks | Targeted four sites on Chr21, using size selection to enrich smaller fetal DNA fragments | T21: sensitivity = 100%; overall accuracy = 99.7% | No direct FF measurement |
| Tan C et al. (2019) [105] | T21, T18 | ddPCR | Cut-off value: normal = 30; validation: normal = 26, T21 = 4; GA: 12–25+2 weeks | Detected ratios of Chr21/18 with 20 loci probes, using LNA probes for better accuracy | Accuracy = 100% | No T18 validation; no direct FF measurement |
| Haidong W et al. (2020) [106] | T21, T18, T13 | ddPCR | Cut-off value: normal = 50, T21 = 5, T18 = 2, T13 = 1; validation: normal = 201, T21 = 10; GA: 11–27 weeks | The iSAFE NIPT assay detected T21,18,13 in a single ddPCR reaction and can be implemented in decentralized labs, offering a rapid solution within 2.5 h | T21: sensitivity = 100%; specificity = 100% | No T18 or T13 validation samples; no direct FF measurement |
| Chen X et al. (2021) [107] | T21 | ddPCR | Normal = 13, T21 = 2; GA: 14–20 weeks | A computational program was used to design highly specific primers and probes targeting SD on Chr21, for the detection of T21 using ddPCR | T21: accuracy = 100% | ddPCR complexity requires 8 pre-amplifications and 8 reactions; SD limitation: SNPs/CNVs affect amplification |
| Ramesh M et al. (2023) [108] | Aneuploidies in Chr13,18,21,22,X,Y | ddPCR | NA | A 120-plex assay for aneuploidy detection and a 60-plex assay for fetal fraction quantification successfully detected chromosomal aneuploidies at fetal fractions as low as 4% | Demonstrated strong concordance with NGS, although no specific accuracy rate was provided | NA |
| Dai P et al. (2022) [73] | T21, T18, T13 | ddPCR | Cut-off value: normal = 170; validation: normal = 247, T21 = 25, T18 = 10, T13 = 1; GA: 12–36 weeks | 10 sets of primers and probes were used for Chr21,18,13, with dPCR calculating ratios in reference to each other | Sensitivity = 100%; specificity = 95% | High cfDNA input requirement (≥0.2 ng/μL cfDNA); no clear cost analysis |
| Lassakova S et al. (2023) [109] | T21 | ddPCR | Cut-off value: normal = 26, T21 = 16; validation: normal = 24, T21 = 6; GA: 13–18 weeks | 16 amplicons from Chr21 and Chr18 (as a reference), with 2 LNA probes to accurately detect reaction products | Sensitivity = 100%; specificity = 100% | Reaction complexity (12/sample): high droplet count (~240 K) for accuracy |
| Author (Year) | Focus of Study | Method Used | Sample Information | Key Findings/Implications | Diagnostic Accuracy |
|---|---|---|---|---|---|
| Wang J et al. (2023) [115] | 22q11.2 deletion/duplication syndrome | cdPCR | Normal = 115; duplication = 9; deletion = 6; GA: 17+1–27 weeks | Six detection sites in the 22q11.2 region A-D were targeted, using z-scores to differentiate normal from affected samples by comparing copy number ratios | Sensitivity = 73.3%; specificity = 96.5%; PPV = 73.3%; NPV = 96.5% |
| Author (Year) | Focus of Study | Method Used | Sample Information | Key Findings/Implications | Diagnostic Accuracy | Reason/Potential Risk for Low Specificity |
|---|---|---|---|---|---|---|
| Charoenkwan P et al. (2022) [123] | β-thalassemia | cdPCR | 35 carriers at risk of having severe β-thalassemia fetuses; GA: 12–18 weeks | The MIB-M/MIB-N ratio was effectively used to differentiate between fetal and maternal DNA | For PIB: sensitivity = 100%; for MIB: sensitivity = 100%; specificity = 92.3% | Maternal DNA interference; low FF; overlapping of MIB-M/MIB-N ratios |
| Sawakwongpra K et al. (2021) [124] | α and β-thalassemia | ddPCR | 46 carriers (22 cases with SEA deletion, 16 cases with HbE, 8 cases with CD41/42 mutation); GA: 17–27 weeks | High accuracy for α-thalassemia; less reliable for β-thalassemia | For SEA deletion: sensitivity = 95.4%, specificity = 91.0%; for HbE: 10 correct, 3 inconclusive, 3 misclassified; for CD41/42 mutation: 2 correct, 4 inconclusive, 2 misclassified | For SEA: low FF (3%) and cfDNA instability in SEA region; for CD41/42 mutation: high ddPCR variability, low positive droplet count, poor probe binding |
| Suwannakhon N et al. (2023) [125] | β-thalassemia | ddPCR | 42 carriers with common mutations (CD41/42, CD17, IVS1-1, CD26); GA: 7–16 weeks | Negative PIB indicates that the fetus is unaffected; positive PIB but negative MIB indicates that the fetus is heterozygous; positive PIB and positive MIB indicates that there is an over-representation of MIBs, and the fetus has compound heterozygous β-thalassemia | 100% concordance with those of amniocentesis | Potential false-positive risks: maternal DNA interference; ddPCR allelic imbalance; low cffDNA concentration |
| D’Aversa E. et al. (2022) [126] | β-thalassemia | ddPCR | 52 maternal plasma samples (PIB = 23, β+IVSI-110/N, β039/N; MIB = 30, heterozygous N/M mothers; homozygous β+IVSI-110/β+IVSI-110 fetus = 1); GA: 5–39 weeks | Identified paternally inherited mutations in 23 samples; M/N allelic ratio used to distinguish fetal genotypes for maternally inherited mutations | Classified 51 of 52 samples correctly | M/N ratio at the boundary; limitations of the z-score classification method; statistical method errors |
| Barrett AN et al. (2012) [127] | Sickle cell anemia | cdPCR | 65 maternal plasma samples (45 male and 20 female fetuses); GA: 11+3–16+5 weeks | The RMD method detected mutations; in female fetuses, indel markers were informative in 65% of cases; allelic ratio analysis distinguished homozygous from heterozygous cases | The classification rate was 82% for male fetuses and 75% for female fetuses; with fetal DNA ≥ 7%, dPCR accuracy = 100% | Delays processing time; indel markers for female fetuses are less effective; long amplicons reduce DNA measurement accuracy; DYS14 copy number differences cause errors |
| Tsui NB et al. (2011) [128] | Hemophilia A and B | mdPCR | 12 samples from 7 hemophilia carriers with male fetuses (hemophilia A = 3, hemophilia B = 4), 20 samples from non-carriers with healthy male fetuses; GA: ≥11 weeks | The RMD method combined with dPCR accurately detected hemophilia A and B in male fetuses as early as 11 weeks of gestation | The fetal genotypes in the 12 plasma samples were detected by dPCR and were found to be consistent with the classifications by the SPRT algorithm | Potential false-positive risks: PCR probe cross-hybridization; cffDNA fraction below 10%; SPRT method’s inability to accurately classify borderline cases |
| Hudecova I et al. (2017) [129] | Hemophilia A and B | ddPCR | 15 carriers of F8 or F9 gene variants; GA: 8–42 weeks | Family-specific assays targeted F8/F9 mutations; ZFY/ZFX assays determined fetal sex and DNA; RMD with SPRT classified hemophilia status by allele balance | In 15 pregnancies, 12 were accurately determined, and 3 were unclassified, but no misclassifications occurred | For 3 unclassified: 2 had low FF (0.8%, 4.0%); 1 had much lower total DNA; SPRT failed due to low fetal DNA or few wells |
| Orhant L et al. (2016) [130] | Achondroplasia | ddPCR | 26 samples from women at risk of fetal achondroplasia, 2 samples from normal women and fetuses; GA: third trimester | The combination of ddPCR and mini-sequencing can accurately detect single-point mutations (c.1138G>A and c.1138G>C) in the FGFR3 gene from maternal plasma | Sensitivity = 100%; specificity = 100% (95% CI, 84.5–100%) | Potential false-positive risks: low cfDNA fragmentation; competition with maternal DNA; low FF |
| Pacault M et al. (2022) [131] | Achondroplasia, thanatophoric dysplasia (TD), common mutations of the FGFR3 gene, neurofibromatosis type 1 (NF1), and cystic fibrosis (CF) | ddPCR | 202 tests from 175 families at risk for single-gene disorders (achondroplasia = 54, TD = 1, all common mutations of the FGFR3 gene = 4, CF = 69, NF1 = 24); GA: ≥8 weeks | ddPCR detected specific FGFR3 gene mutations linked to achondroplasia (c.1138G>A, c.1138G>C) and TD (c.742C>T, c.1118A>G, c.1948A>G) from maternal plasma; ddPCR distinguished closely located CFTR mutations (c.1519_1521del and c.1521_1523del), overcoming the challenge of genomic proximity; assays for NF1 were designed to distinguish between wild-type and mutant alleles for the identification and quantification of paternal NF1 mutations | For achondroplasia, TD, and FGFR3 gene mutation: 19 cases had the FGFR3 c.1138G>A mutation, 1 had c.1138G>C, and no TD mutations; results fully consistent with invasive prenatal testing and no inconclusive outcomes; for CF: 56% of samples were detected as paternal mutation, 1 case was inconclusive, and results were completely consistent with those of invasive prenatal tests; for NF1: 1 assay for c.2033dup could not be designed | For CF: egg donor’s genetic status was unknown; for NF1: technical limitations caused by a polyC region |
| Wei X et al. (2020) [78] | SMA (SMN1) | ddPCR | Set A: 17 SMA carriers with male fetuses, Set B: randomly selected 10 women from Set A and analyzed under blinding; GA: 16–22 weeks | The 6th nucleotide of SMN1 exon 7 was targeted, enabling precise detection of SMN1 deletions and SMN1-to-SMN2 conversions, both major causes of SMA | The concordance rates with MLPA for Set A and Set B were 94.1% and 90%, respectively, and in all classifiable tests, ddPCR achieved 100% concordance with MLPA | Low FF and concentration |
| Chang MY et al. (2018) [132] | Hereditary hearing loss | Picodroplet dPCR and cdPCR (used separately) | 3 families with known autosomal recessive mutations (GJB2 c.235delC, SLC26A4 IVS7-2A>G); GA: 16–27 weeks | Chi-squared and Bayesian analysis predicted fetal genotypes using mutant allele proportions, bypassing the need for fetal DNA fraction or paternal SNPs | Successfully predicted fetal genotypes in all families with high accuracy using both dPCR methods | Potential false-positive risks: borderline mutant allele frequencies; maternal DNA control limitations; inherent dPCR error rates |
| Gruber A et al. (2018) [133] | NF1 and CF | ddPCR | 8 families (NF1 = 4, CF = 4); GA: 8–15 weeks | Identified paternal mutations in NF1 and CFTR mutations | Paternal mutation results were completely consistent with those of invasive prenatal tests | Potential false-positive risks: due to sequence complexity or probe issues; challenges in rare event detection |
| Debrand E et al. (2015) [134] | CF | ddPCR | 1 couple (3 pregnancies) carrying different mutated CFTR alleles, 6 normal; GA: 11–12 weeks | Exon 11 of the CFTR gene was targeted to quantify the mutant (ΔF508-MUT; FAM) and normal (ΔF508-NOR; VIC) alleles at position c.1521_1523, enabling the detection of paternal CFTR mutations | The ΔF508 CFTR mutant allele was correctly identified in the three fetuses affected by CF, and it was not detected in the six control fetuses; consistent with traditional invasive testing | Potential false-positive risks: droplet carry-over contamination; low background noise |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Charoenkwan, P.; Traisrisilp, K.; Piyamongkol, W.; Tongprasert, F. Application of Digital Polymerase Chain Reaction (dPCR) in Non-Invasive Prenatal Testing (NIPT). Biomolecules 2025, 15, 360. https://doi.org/10.3390/biom15030360
Guo Y, Charoenkwan P, Traisrisilp K, Piyamongkol W, Tongprasert F. Application of Digital Polymerase Chain Reaction (dPCR) in Non-Invasive Prenatal Testing (NIPT). Biomolecules. 2025; 15(3):360. https://doi.org/10.3390/biom15030360
Chicago/Turabian StyleGuo, Ying, Pimlak Charoenkwan, Kuntharee Traisrisilp, Wirawit Piyamongkol, and Fuanglada Tongprasert. 2025. "Application of Digital Polymerase Chain Reaction (dPCR) in Non-Invasive Prenatal Testing (NIPT)" Biomolecules 15, no. 3: 360. https://doi.org/10.3390/biom15030360
APA StyleGuo, Y., Charoenkwan, P., Traisrisilp, K., Piyamongkol, W., & Tongprasert, F. (2025). Application of Digital Polymerase Chain Reaction (dPCR) in Non-Invasive Prenatal Testing (NIPT). Biomolecules, 15(3), 360. https://doi.org/10.3390/biom15030360