
The Progress and Evolving Trends in Nucleic-Acid-Based Therapies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
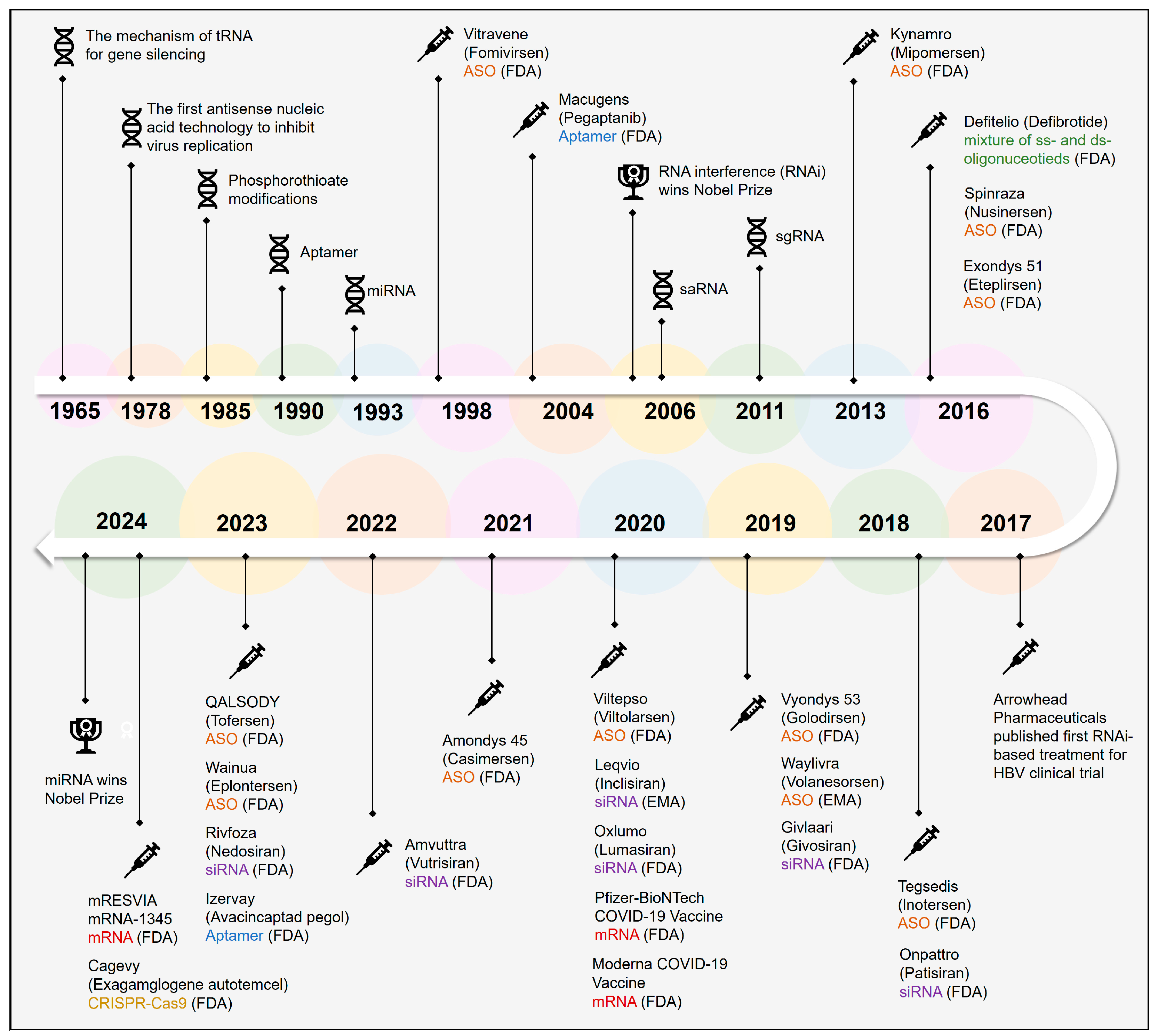
:1. Introduction
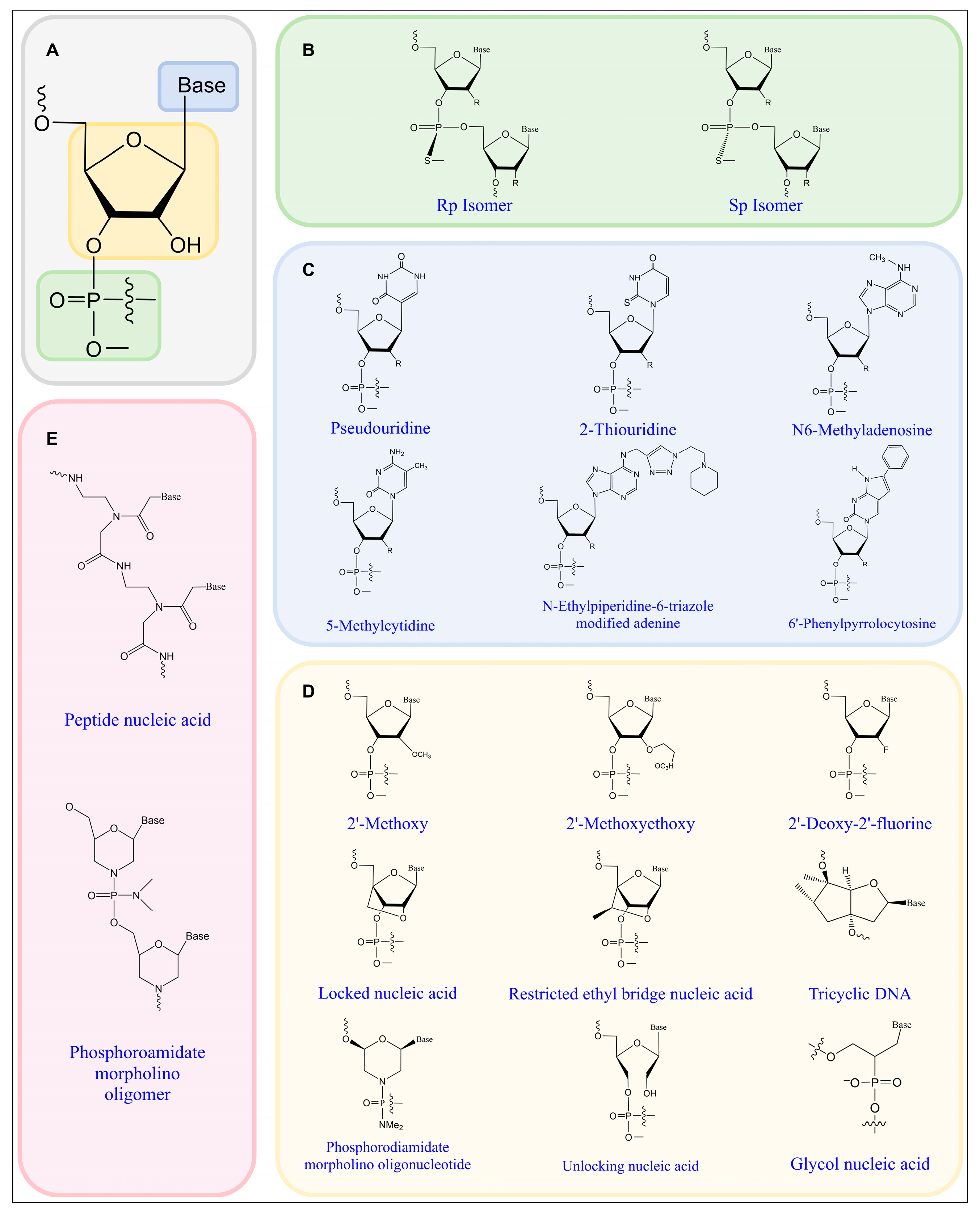
2. Chemical Modifications of Nucleic-Acid-Based Therapeutics
2.1. Phosphate Backbone Modifications
2.2. Base Modifications
2.3. Ribose Modifications
2.4. Other Specialized Modifications
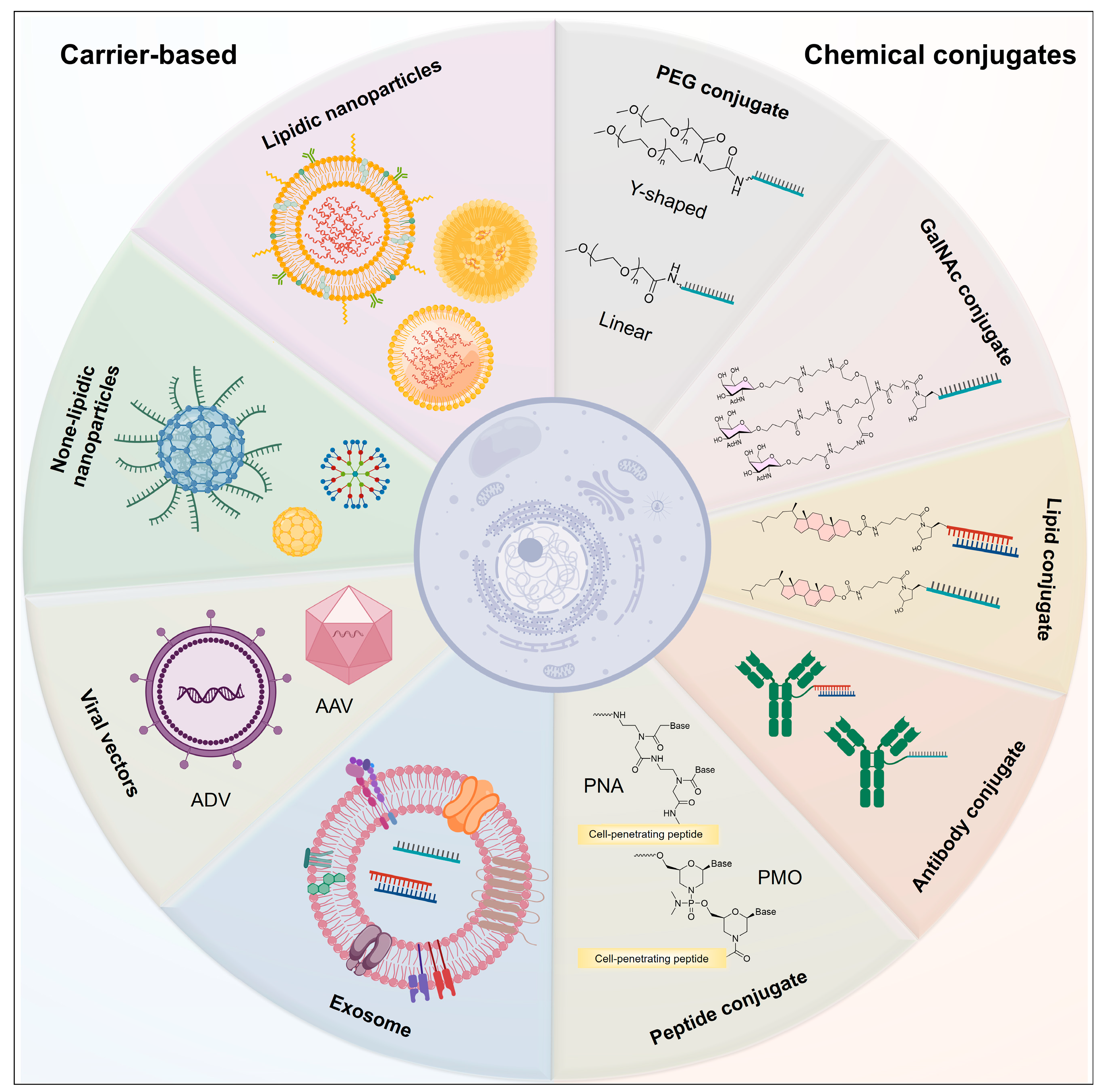
3. Delivery Strategies for Nucleic Acid Drugs
3.1. PEG Conjugates
3.2. Lipid-Conjugated Oligonucleotide
3.3. GalNAc Conjugates
3.4. Antibody-Conjugated Oligonucleotide
3.5. Peptide Conjugates
3.6. Lipidic Nanoparticles
3.7. None-Lipidic Nanoparticles
3.8. Exosomes
4. Antisense Oligonucleotides (ASOs)

5. Small Interfering RNAs (siRNAs)
6. MicroRNAs (miRNAs)
7. Aptamer RNA
8. Messenger RNA (mRNA)
9. CRISPR-Cas9 Gene Editing System
10. Clinical Applications of Nucleic Acid Drugs
11. Frontiers and Prospects
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- SoRelle, R. Who owns your DNA? Who will own it? Circulation 2000, 101, e67–e68. [Google Scholar] [CrossRef] [PubMed]
- Lächelt, U.; Wagner, E. Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar] [CrossRef] [PubMed]
- Belgrad, J.; Fakih, H.H.; Khvorova, A. Nucleic Acid Therapeutics: Successes, Milestones, and Upcoming Innovation. Nucleic Acid Ther. 2024, 34, 52–72. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Abdullah, R.; Wu, X.; Li, F.; Ma, Y.; Lu, A.; Zhang, G. Pancreatic cancer: Nucleic acid drug discovery and targeted therapy. Front. Cell Dev. Biol. 2022, 10, 855474. [Google Scholar] [CrossRef]
- Rosie, Z.Y.; Lemonidis, K.M.; Graham, M.J.; Matson, J.E.; Crooke, R.M.; Tribble, D.L.; Wedel, M.K.; Levin, A.A.; Geary, R.S. Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem. pharmacol. 2009, 77, 910–919. [Google Scholar]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef]
- Sands, H.; Gorey-Feret, L.J.; Cocuzza, A.J.; Hobbs, F.W.; Chidester, D.; Trainor, G.L. Biodistribution and metabolism of internally 3H-labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol. Pharmacol. 1994, 45, 932–943. [Google Scholar] [CrossRef]
- Stoddard, B.L.; Khvorova, A.; Corey, D.R.; Dynan, W.S.; Fox, K.R. Nucleic Acids Research and Nucleic Acid Therapeutics. Nucleic Acids Res. 2018, 46, 1563–1564. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Liu, L.; Gao, H.; Guo, C.; Liu, T.; Li, N.; Qian, Q. Therapeutic mechanism of nucleic acid drugs. ChemistrySelect 2021, 6, 903–916. [Google Scholar] [CrossRef]
- Plescia, O.J.; Palczuk, N.C.; Cora-Figueroa, E.; Mukherjee, A.; Braun, W. Production of antibodies to soluble RNA (sRNA). Proc. Natl. Acad. Sci. USA 1965, 54, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, L.; Li, M.; Tian, Z.; Yang, M.; Wu, X.; Wang, X.; Shang, G.; Xie, M.; Chen, Y.; et al. Structures of human TR4LBD-JAZF1 and TR4DBD-DNA complexes reveal the molecular basis of transcriptional regulation. Nucleic Acids Res. 2023, 51, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Marwick, C. First antisense drug will treat CMV retinitis. JAMA 1998, 280, 871. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham Jr, E.T.; Feinsod, M.; Guyer, D.R. Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef]
- Li, L.-C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van Der Oost, J.; Brouns, S.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119. [Google Scholar]
- Kornblum, N.; Ayyanar, K.; Benimetskaya, L.; Richardson, P.; Iacobelli, M.; Stein, C. Defibrotide, a polydisperse mixture of single-stranded phosphodiester oligonucleotides with lifesaving activity in severe hepatic veno-occlusive disease: Clinical outcomes and potential mechanisms of action. Oligonucleotides 2006, 16, 105–114. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for spinal muscular atrophy. Ther. Adv. Neurol. Disord. 2018, 11, 1756285618754459. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.-F.; Chan, H.L.-Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First global approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Syed, Y.Y. Givosiran: A review in acute hepatic porphyria. Drugs 2021, 81, 841–848. [Google Scholar] [CrossRef]
- Heo, Y.-A. Golodirsen: First approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S. Volanesorsen: First global approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Viltolarsen: First approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Inclisiran: First approval. Drugs 2021, 81, 389–395. [Google Scholar] [CrossRef]
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; van’t Hoff, W.G. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N. Engl. J. Med. 2021, 384, 1216–1226. [Google Scholar] [CrossRef]
- Ledford, H. Moderna COVID vaccine becomes second to get US authorization. Nature 2020. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Keam, S.J. Vutrisiran: First approval. Drugs 2022, 82, 1419–1425. [Google Scholar] [CrossRef]
- Jin, J.; Zhong, X.-B. ASO drug Qalsody (tofersen) targets amyotrophic lateral sclerosis. Trends Pharmacol. Sci. 2023, 44, 1043–1044. [Google Scholar] [CrossRef]
- Wang, J.; Tan, M.; Wang, Y.; Liu, X.; Lin, A. Advances in modification and delivery of nucleic acid drugs. Zhejiang Da Xue Xue Bao Yi Xue Ban 2023, 52, 417–428. [Google Scholar] [CrossRef]
- Syed, Y.Y. Nedosiran: First approval. Drugs 2023, 83, 1729–1733. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Avacincaptad pegol: First approval. Drugs 2023, 83, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Orders, M. FDA authorizes Pfizer-BioNTech COVID-19 vaccine. Med. Lett. Drugs Ther. 2021, 63, 1–2. [Google Scholar]
- Hoy, S.M. Exagamglogene Autotemcel: First Approval. Mol. Diagn. Ther. 2024, 28, 133–139. [Google Scholar] [CrossRef]
- Britton, A.; Roper, L.E.; Kotton, C.N.; Hutton, D.W.; Fleming-Dutra, K.E.; Godfrey, M.; Ortega-Sanchez, I.R.; Broder, K.R.; Talbot, H.K.; Long, S.S.; et al. Use of Respiratory Syncytial Virus Vaccines in Adults Aged ≥60 Years: Updated Recommendations of the Advisory Committee on Immunization Practices-United States, 2024. MMWR Morb. Mortal. Wkly. Rep. 2024, 73, 696–702. [Google Scholar] [CrossRef]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2019, 25, 3. [Google Scholar] [CrossRef]
- Ni, S.; Zhuo, Z.; Pan, Y.; Yu, Y.; Li, F.; Liu, J.; Wang, L.; Wu, X.; Li, D.; Wan, Y. Recent progress in aptamer discoveries and modifications for therapeutic applications. ACS Appl. Mater. Interfaces 2020, 13, 9500–9519. [Google Scholar] [CrossRef]
- Prakash, T.P. An overview of sugar-modified oligonucleotides for antisense therapeutics. Chem. Biodivers. 2011, 8, 1616–1641. [Google Scholar] [CrossRef]
- Raoof, A.A.; Ramtoola, Z.; McKenna, B.; Yu, R.Z.; Hardee, G.; Geary, R.S. Effect of sodium caprate on the intestinal absorption of two modified antisense oligonucleotides in pigs. Eur. J. Pharm. Sci. 2002, 17, 131–138. [Google Scholar] [CrossRef]
- Iwamoto, N.; Butler, D.C.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.; Meena; Standley, S.M.; Lu, G.; Apponi, L.H. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol 2017, 35, 845–851. [Google Scholar] [CrossRef]
- De Smet, M.D.; Meenken, C.; van den Horn, G.J. Fomivirsen–a phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflamm. 1999, 7, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.; Porteus, M.H. Uridine depletion and chemical modification increase Cas9 mRNA activity and reduce immunogenicity without HPLC purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, R.A.P.; Suter, S.R.; Ball-Jones, A.A.; Ibarra-Soza, J.M.; Zheng, Y.; Beal, P.A. Base modification strategies to modulate immune stimulation by an siRNA. ChemBioChem. 2015, 16, 262–267. [Google Scholar] [CrossRef]
- Wahba, A.S.; Azizi, F.; Deleavey, G.F.; Brown, C.; Robert, F.; Carrier, M.; Kalota, A.; Gewirtz, A.M.; Pelletier, J.; Hudson, R.H. Phenylpyrrolocytosine as an unobtrusive base modification for monitoring activity and cellular trafficking of siRNA. ACS Chem. Biol. 2011, 6, 912–919. [Google Scholar] [CrossRef]
- Shen, W.; Liang, X.-h.; Sun, H.; Crooke, S.T. 2′-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 2015, 43, 4569–4578. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Warren, T.K.; Shurtleff, A.C.; Bavari, S. Advanced morpholino oligomers: A novel approach to antiviral therapy. Antivir. Res. 2012, 94, 80–88. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new? A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef]
- Osborn, M.F.; Coles, A.H.; Biscans, A.; Haraszti, R.A.; Roux, L.; Davis, S.; Ly, S.; Echeverria, D.; Hassler, M.R.; Godinho, B.; et al. Hydrophobicity drives the systemic distribution of lipid-conjugated siRNAs via lipid transport pathways. Nucleic Acids Res. 2019, 47, 1070–1081. [Google Scholar] [CrossRef]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Østergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Wang, S.; Allen, N.; Prakash, T.P.; Liang, X.-h.; Crooke, S.T. Lipid Conjugates Enhance Endosomal Release of Antisense Oligonucleotides into Cells. Nucleic Acid Ther. 2019, 29, 245–255. [Google Scholar] [CrossRef]
- Tanowitz, M.; Hettrick, L.; Revenko, A.; Kinberger, G.A.; Prakash, T.P.; Seth, P.P. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017, 45, 12388–12400. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Yu, R.Z.; Gaus, H.J.; Greenlee, S.; Post, N.; Schmidt, K.; Migawa, M.T.; Seth, P.P.; Zanardi, T.A.; Prakash, T.P.; et al. Elucidation of the Biotransformation Pathways of a Galnac3-conjugated Antisense Oligonucleotide in Rats and Monkeys. Mol. Ther. Nucleic Acids 2016, 5, e319. [Google Scholar] [CrossRef]
- Huang, Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Mol. Ther. Nucleic Acids 2017, 6, 116–132. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, R.Z.; Henry, S.; Geary, R.S. Pharmacokinetics and Clinical Pharmacology Considerations of GalNAc(3)-Conjugated Antisense Oligonucleotides. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 475–485. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Xia, S.; Yu, R.Z.; Viney, N.J.; Wang, Y.; Tsimikas, S.; Geary, R.S. Integrated Assessment of the Clinical Performance of GalNAc(3)-Conjugated 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides: I. Human Volunteer Experience. Nucleic Acid Ther. 2019, 29, 16–32. [Google Scholar] [CrossRef]
- Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P.P.; Li, Y.; Beckett, L.; et al. Novel Targeted Therapy for Precursor B Cell Acute Lymphoblastic Leukemia: Anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632–642. [Google Scholar] [CrossRef]
- Arnold, A.E.; Malek-Adamian, E.; Le, P.U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.; Damha, M.J.; Shoichet, M.S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a Key Gene in Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 518–527. [Google Scholar] [CrossRef]
- Ibtehaj, N.; Bahauddin, A.; Ivannikov, M.; Rytting, E.; Jamaluddin, M.; Liang, Y.; Sun, J.; Haller, S.L.; Wu, X.; Huda, R. B cell-specific mAb–siRNA conjugates improve experimental myasthenia. J. Autoimmun. 2023, 135, 102983. [Google Scholar] [CrossRef]
- Ibtehaj, N.; Huda, R. High-dose BAFF receptor specific mAb-siRNA conjugate generates Fas-expressing B cells in lymph nodes and high-affinity serum autoantibody in a myasthenia mouse model. Clin. Immunol. 2017, 176, 122–130. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control Release 2016, 237, 1–13. [Google Scholar] [CrossRef]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J.A. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019, 29, 1–12. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef]
- Liao, L.; Cen, B.; Li, G.; Wei, Y.; Wang, Z.; Huang, W.; He, S.; Yuan, Y.; Ji, A. A bivalent cyclic RGD–siRNA conjugate enhances the antitumor effect of apatinib via co-inhibiting VEGFR2 in non-small cell lung cancer xenografts. Drug Deliv. 2021, 28, 1432–1442. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Hu, M.; Li, X.; You, Z.; Cai, R.; Chen, C. Physiological Barriers and Strategies of Lipid-Based Nanoparticles for Nucleic Acid Drug Delivery. Adv. Mater. 2024, 36, 2303266. [Google Scholar] [CrossRef]
- Ku, S.H.; Jo, S.D.; Lee, Y.K.; Kim, K.; Kim, S.H. Chemical and structural modifications of RNAi therapeutics. Adv. Drug Deliv. Rev. 2016, 104, 16–28. [Google Scholar] [CrossRef]
- Jung, H.N.; Lee, S.Y.; Lee, S.; Youn, H.; Im, H.J. Lipid nanoparticles for delivery of RNA therapeutics: Current status and the role of in vivo imaging. Theranostics 2022, 12, 7509–7531. [Google Scholar] [CrossRef]
- Zhang, L.; More, K.R.; Ojha, A.; Jackson, C.B.; Quinlan, B.D.; Li, H.; He, W.; Farzan, M.; Pardi, N.; Choe, H. Effect of mRNA-LNP components of two globally-marketed COVID-19 vaccines on efficacy and stability. NPJ Vaccines 2023, 8, 156. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; van der Meel, R.; Zaifman, J.; Darjuan, M.M.; Grisch-Chan, H.M.; Thöny, B.; Tam, Y.Y.C.; Cullis, P.R. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 2019, 11, 9023–9031. [Google Scholar] [CrossRef]
- Ferraresso, F.; Strilchuk, A.W.; Juang, L.J.; Poole, L.G.; Luyendyk, J.P.; Kastrup, C.J. Comparison of DLin-MC3-DMA and ALC-0315 for siRNA Delivery to Hepatocytes and Hepatic Stellate Cells. Mol. Pharm. 2022, 19, 2175–2182. [Google Scholar] [CrossRef]
- Ke, X.; Shelton, L.; Hu, Y.; Zhu, Y.; Chow, E.; Tang, H.; Santos, J.L.; Mao, H.Q. Surface-Functionalized PEGylated Nanoparticles Deliver Messenger RNA to Pulmonary Immune Cells. ACS Appl. Mater. Interfaces 2020, 12, 35835–35844. [Google Scholar] [CrossRef]
- Rai, R.; Alwani, S.; Badea, I. Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications. Polymers 2019, 11, 745. [Google Scholar] [CrossRef]
- Yang, W.; Chen, P.; Boonstra, E.; Hong, T.; Cabral, H. Polymeric Micelles with pH-Responsive Cross-Linked Core Enhance In Vivo mRNA Delivery. Pharmaceutics 2022, 14, 1205. [Google Scholar] [CrossRef]
- Djafari, J.; Fernández-Lodeiro, J.; Santos, H.M.; Lorenzo, J.; Rodriguez-Calado, S.; Bértolo, E.; Capelo-Martínez, J.L.; Lodeiro, C. Study and Preparation of Multifunctional Poly(L-Lysine)@Hyaluronic Acid Nanopolyplexes for the Effective Delivery of Tumor Suppressive MiR-34a into Triple-Negative Breast Cancer Cells. Materials 2020, 13, 5309. [Google Scholar] [CrossRef]
- Souri, M.; Bagherzadeh, M.A.; Mofazzal Jahromi, M.A.; Mohammad-Beigi, H.; Abdoli, A.; Mir, H.; Roustazadeh, A.; Pirestani, M.; Sahandi Zangabad, P.; Kiani, J.; et al. Poly-L-lysine/hyaluronan nanocarriers as a novel nanosystem for gene delivery. J. Microsc. 2022, 287, 32–44. [Google Scholar] [CrossRef]
- Luther, D.C.; Huang, R.; Jeon, T.; Zhang, X.; Lee, Y.W.; Nagaraj, H.; Rotello, V.M. Delivery of drugs, proteins, and nucleic acids using inorganic nanoparticles. Adv. Drug Deliv. Rev. 2020, 156, 188–213. [Google Scholar] [CrossRef]
- Kumthekar, P.; Ko, C.H.; Paunesku, T.; Dixit, K.; Sonabend, A.M.; Bloch, O.; Tate, M.; Schwartz, M.; Zuckerman, L.; Lezon, R.; et al. A first-in-human phase 0 clinical study of RNA interference-based spherical nucleic acids in patients with recurrent glioblastoma. Sci. Transl. Med. 2021, 13, eabb3945. [Google Scholar] [CrossRef]
- Jensen, S.A.; Day, E.S.; Ko, C.H.; Hurley, L.A.; Luciano, J.P.; Kouri, F.M.; Merkel, T.J.; Luthi, A.J.; Patel, P.C.; Cutler, J.I.; et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [Google Scholar] [CrossRef]
- Rahamathulla, M.; Bhosale, R.R.; Osmani, R.A.M.; Mahima, K.C.; Johnson, A.P.; Hani, U.; Ghazwani, M.; Begum, M.Y.; Alshehri, S.; Ghoneim, M.M.; et al. Carbon Nanotubes: Current Perspectives on Diverse Applications in Targeted Drug Delivery and Therapies. Materials 2021, 14, 6707. [Google Scholar] [CrossRef]
- Chavan, N.; Dharmaraj, D.; Sarap, S.; Surve, C. Magnetic nanoparticles–A new era in nanotechnology. J. Drug Deliv. Sci. Technol. 2022, 77, 103899. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Zhang, X.; Huang, H.; Tang, S.; Chai, Y.; Xu, Z.; Li, M.; Chen, X.; Liu, J.; et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J. Nanobiotechnol. 2022, 20, 279. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Desai, N.N.; Qureshi, M.Z.; Librelotto, D.R.N.; Gasparri, M.L.; Bishayee, A.; Nabavi, S.M.; Curti, V.; Daglia, M. Exosome biogenesis, bioactivities and functions as new delivery systems of natural compounds. Biotechnol. Adv. 2018, 36, 328–334. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; La Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Control. Release 2017, 266, 8–16. [Google Scholar] [CrossRef]
- Yang, Z.; Shi, J.; Xie, J.; Wang, Y.; Sun, J.; Liu, T.; Zhao, Y.; Zhao, X.; Wang, X.; Ma, Y.; et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Hu, B.; Li, B.; Li, K.; Liu, Y.; Li, C.; Zheng, L.; Zhang, M.; Yang, T.; Guo, S.; Dong, X.; et al. Thermostable ionizable lipid-like nanoparticle (iLAND) for RNAi treatment of hyperlipidemia. Sci. Adv. 2022, 8, eabm1418. [Google Scholar] [CrossRef]
- Gong, C.; Zhang, X.; Shi, M.; Li, F.; Wang, S.; Wang, Y.; Wang, Y.; Wei, W.; Ma, G. Tumor Exosomes Reprogrammed by Low pH Are Efficient Targeting Vehicles for Smart Drug Delivery and Personalized Therapy against their Homologous Tumor. Adv. Sci. 2021, 8, 2002787. [Google Scholar] [CrossRef]
- Kim, G.; Kim, M.; Lee, Y.; Byun, J.W.; Hwang, D.W.; Lee, M. Systemic delivery of microRNA-21 antisense oligonucleotides to the brain using T7-peptide decorated exosomes. J. Control. Release 2020, 317, 273–281. [Google Scholar] [CrossRef]
- Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. [Google Scholar] [CrossRef]
- Couch, Y.; Buzàs, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Théry, C.; et al. A brief history of nearly EV-erything-The rise and rise of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef]
- Angrish, N.; Khare, G. Antisense oligonucleotide based therapeutics and its applications against bacterial infections. Medicine in Drug Discov. 2023, 20, 100166. [Google Scholar] [CrossRef]
- Ringold, S.; Tomlinson, G.; Schanberg, L.; Del Gaizo, V.; Murphy, K.; Feldman, B.; Ong, M.; Natter, M.; Kimura10, Y. Rheumatology Congress. Pediatr. Rheumatol. Online J. 2023, 21, 122. [Google Scholar]
- Kiełpiński, Ł.J.; Hagedorn, P.H.; Lindow, M.; Vinther, J. RNase H sequence preferences influence antisense oligonucleotide efficiency. Nucleic Acids Res. 2017, 45, 12932–12944. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.-h. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Heendeniya, S.N.; Le, B.T.; Rahimizadeh, K.; Rabiee, N.; Zahra, Q.U.A.; Veedu, R.N. Splice-Modulating Antisense Oligonucleotides as Therapeutics for Inherited Metabolic Diseases. Biodrugs 2024, 38, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pan, W.; Xu, Y.; Zhang, J.; Wan, J.; Jiang, H. Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases. J. Inflamm. Res. 2022, 15, 3083–3094. [Google Scholar] [CrossRef] [PubMed]
- Pagnan, G.; Stuart, D.D.; Pastorino, F.; Raffaghello, L.; Montaldo, P.G.; Allen, T.M.; Calabretta, B.; Ponzoni, M. Delivery of c-myb antisense oligodeoxynucleotides to human neuroblastoma cells via disialoganglioside GD(2)-targeted immunoliposomes: Antitumor effects. J. Nat. Cancer Institute 2000, 92, 253–261. [Google Scholar] [CrossRef]
- Sicard, G.; Paris, C.; Giacometti, S.; Rodallec, A.; Ciccolini, J.; Rocchi, P.; Fanciullino, R. Enhanced Antisense Oligonucleotide Delivery Using Cationic Liposomes Grafted with Trastuzumab: A Proof-of-Concept Study in Prostate Cancer. Pharmaceutics 2020, 12, 1166. [Google Scholar] [CrossRef]
- Hughes, J.; Astriab, A.; Yoo, H.; Alahari, S.; Liang, E.; Sergueev, D.; Shaw, B.R.; Juliano, R.L. In vitro transport and delivery of antisense oligonucleotides. Methods Enzymol. 2000, 313, 342–358. [Google Scholar]
- Lysik, M.A.; Pong, S.W. Innovations in oligonucleotide drug delivery. J. Pharm. Sci. 2003, 92, 1559–1573. [Google Scholar] [CrossRef]
- Thomas, G.S.; Cromwell, W.C.; Ali, S.; Chin, W.; Flaim, J.D.; Davidson, M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: A randomized, double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2013, 62, 2178–2184. [Google Scholar] [CrossRef]
- Van Roon-Mom, W.; Ferguson, C.; Aartsma-Rus, A. From Failure to Meet the Clinical Endpoint to U.S. Food and Drug Administration Approval: 15th Antisense oligonucleotide therapy approved qalsody (Tofersen) for treatment of SOD1 mutated amyotrophic lateral sclerosis. Nucleic Acid Ther. 2023, 33, 234–237. [Google Scholar] [CrossRef]
- Wang, F.; Zuroske, T.; Watts, J.K. RNA therapeutics on the rise. Nat. Rev. Drug Discov. 2020, 19, 441–442. [Google Scholar] [CrossRef]
- Jansson-Löfmark, R.; Gennemark, P. Inferring Half-Lives at the Effect Site of Oligonucleotide Drugs. Nucleic Acid Ther. 2018, 28, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, H.; Zhang, L.; Mu, W.; Zhang, Y.; Chen, T.; Wu, J.; Tang, H.; Zheng, S.; Liu, Y.; et al. Generic Diagramming Platform (GDP): A comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 2024, 53, D1670–D1676. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Dasaradhi, P.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA interference: Biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 2003, 67, 657–685. [Google Scholar] [CrossRef] [PubMed]
- Tomari, Y.; Zamore, P.D. Perspective: Machines for RNAi. Genes Dev. 2005, 19, 517–529. [Google Scholar] [CrossRef]
- Claycomb, J.M. Ancient endo-siRNA pathways reveal new tricks. Curr. Biol. 2014, 24, R703–R715. [Google Scholar] [CrossRef]
- Wang, D.; Ruvkun, G. Regulation of Caenorhabditis elegans RNA interference by the daf-2 insulin stress and longevity signaling pathway. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2004; Volume 69, pp. 429–432. [Google Scholar] [CrossRef]
- Fenske, D.B.; Chonn, A.; Cullis, P.R. Liposomal nanomedicines: An emerging field. Toxicol. Pathol. 2008, 36, 21–29. [Google Scholar] [CrossRef]
- Hayes, M.; Drummond, D.; Hong, K.; Park, J.; Marks, J.; Kirpotin, D. Assembly of nucleic acid-lipid nanoparticles from aqueous-organic monophases. Biochim. Biophys. Acta 2006, 1758, 429–442. [Google Scholar] [CrossRef]
- Ambegia, E.; Ansell, S.; Cullis, P.; Heyes, J.; Palmer, L.; MacLachlan, I. Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim. Biophys. Acta 2005, 1669, 155–163. [Google Scholar] [CrossRef]
- Tam, Y.Y.; Chen, S.; Cullis, P.R. Advances in Lipid Nanoparticles for siRNA Delivery. Pharmaceutics 2013, 5, 498–507. [Google Scholar] [CrossRef]
- Zimmermann, T.S.; Lee, A.C.; Akinc, A.; Bramlage, B.; Bumcrot, D.; Fedoruk, M.N.; Harborth, J.; Heyes, J.A.; Jeffs, L.B.; John, M.; et al. RNAi-mediated gene silencing in non-human primates. Nature 2006, 441, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Goldberg, M.; Qin, J.; Dorkin, J.R.; Gamba-Vitalo, C.; Maier, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Manoharan, M.; et al. Development of lipidoid-siRNA formulations for systemic delivery to the liver. Mol. Ther. 2009, 17, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef]
- Kang, H.; Ga, Y.J.; Kim, S.H.; Cho, Y.H.; Kim, J.W.; Kim, C.; Yeh, J.Y. Small interfering RNA (siRNA)-based therapeutic applications against viruses: Principles, potential, and challenges. J. Biomed. Sci. 2023, 30, 88. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef]
- Takahashi, M.; Nagai, C.; Hatakeyama, H.; Minakawa, N.; Harashima, H.; Matsuda, A. Intracellular stability of 2′-OMe-4′-thioribonucleoside modified siRNA leads to long-term RNAi effect. Nucleic Acids Res. 2012, 40, 5787–5793. [Google Scholar] [CrossRef]
- Jahns, H.; Taneja, N.; Willoughby, J.L.; Akabane-Nakata, M.; Brown, C.R.; Nguyen, T.; Bisbe, A.; Matsuda, S.; Hettinger, M.; Manoharan, R.M. Chirality matters: Stereo-defined phosphorothioate linkages at the termini of small interfering RNAs improve pharmacology in vivo. Nucleic Acids Res. 2022, 50, 1221–1240. [Google Scholar] [CrossRef]
- Zu, H.; Gao, D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Singh, S.; Sharma, D.; Webster, T.J.; Shafaat, K.; Faruk, A. Elastic liposomes as novel carriers: Recent advances in drug delivery. Int. J. Nanomed. 2017, 12, 5087–5108. [Google Scholar] [CrossRef] [PubMed]
- Ganju, A.; Khan, S.; Hafeez, B.B.; Behrman, S.W.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. miRNA nanotherapeutics for cancer. Drug Discov. Today 2017, 22, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; How Huang, K.; Jen Lee, M.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Mascini, M.; Palchetti, I.; Tombelli, S. Nucleic acid and peptide aptamers: Fundamentals and bioanalytical aspects. Angew. Chem. Int. Ed. Engl. 2012, 51, 1316–1332. [Google Scholar] [CrossRef]
- Tok, J.B.-H.; Cho, J.; Rando, R.R. RNA aptamers that specifically bind to a 16S ribosomal RNA decoding region construct. Nucleic Acids Res. 2000, 28, 2902–2910. [Google Scholar] [CrossRef]
- Chen, X.; Chamorro, M.; Lee, S.; Shen, L.; Hines, J.; Tinoco Jr, I.; Varmus, H. Structural and functional studies of retroviral RNA pseudoknots involved in ribosomal frameshifting: Nucleotides at the junction of the two stems are important for efficient ribosomal frameshifting. The EMBO J. 1995, 14, 842–852. [Google Scholar] [CrossRef]
- Park, S.-J.; Jung, Y.H.; Kim, Y.-G.; Park, H.-J. Identification of novel ligands for the RNA pseudoknot that regulate−1 ribosomal frameshifting. Bioorg. Med. Chem. 2008, 16, 4676–4684. [Google Scholar] [CrossRef]
- Wilson, C.; Nix, J.; Szostak, J. Functional requirements for specific ligand recognition by a biotin-binding RNA pseudoknot. Biochemistry 1998, 37, 14410–14419. [Google Scholar] [CrossRef] [PubMed]
- Chovelon, B.; Fiore, E.; Faure, P.; Peyrin, E.; Ravelet, C. Mirror-image aptamer kissing complex for arginine-vasopressin sensing. Anal. Chim. Acta 2018, 1001, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Endo, M.; Suzuki, Y.; Hidaka, K.; Durand, G.; Dausse, E.; Toulmé, J.-J.; Sugiyama, H. Single-molecule observations of RNA–RNA kissing interactions in a DNA nanostructure. Biomater. Sci. 2016, 4, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Huang, J.-Y.; Bao, T.; Zhou, J.; Xia, H.-X.; Zhang, X.-H.; Wang, S.-F.; Zhao, Y.-D. Increased electrocatalyzed performance through hairpin oligonucleotide aptamer-functionalized gold nanorods labels and graphene-streptavidin nanomatrix: Highly selective and sensitive electrochemical biosensor of carcinoembryonic antigen. Biosens. Bioelectron. 2016, 83, 142–148. [Google Scholar] [CrossRef]
- Yang, C.; Kulkarni, M.; Lim, M.; Pak, Y. In silico direct folding of thrombin-binding aptamer G-quadruplex at all-atom level. Nucleic Acids Res. 2017, 45, 12648–12656. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, Y.; Gao, L.; Fei, Y.; Ye, T.; Li, Q.; Zhou, X.; Gan, N.; Shao, Y. Structuring polarity-inverted TBA to G-quadruplex for selective recognition of planarity of natural isoquinoline alkaloids. Analyst 2018, 143, 4907–4914. [Google Scholar] [CrossRef]
- Hermann, T.; Patel, D.J. Adaptive recognition by nucleic acid aptamers. Science 2000, 287, 820–825. [Google Scholar] [CrossRef]
- Hoerter, J.A.; Walter, N.G. Chemical modification resolves the asymmetry of siRNA strand degradation in human blood serum. Rna 2007, 13, 1887–1893. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, T.; Romesberg, F.E. Evolved polymerases facilitate selection of fully 2′-OMe-modified aptamers. Chem. Sci. 2017, 8, 8179–8182. [Google Scholar] [CrossRef]
- Awachat, R.; Wagh, A.A.; Aher, M.; Fernandes, M.; Kumar, V.A. Favorable 2′-substitution in the loop region of a thrombin-binding DNA aptamer. Bioorg. Med. Chem. Lett. 2018, 28, 1765–1768. [Google Scholar] [CrossRef]
- London, G.M.; Mayosi, B.M.; Khati, M. Isolation and characterization of 2′-F-RNA aptamers against whole HIV-1 subtype C envelope pseudovirus. Biochem. and Biophys. Res. Commun. 2015, 456, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Svobodova, M.; Bunka, D.; Nadal, P.; Stockley, P.; O’Sullivan, C. Selection of 2′ F-modified RNA aptamers against prostate-specific antigen and their evaluation for diagnostic and therapeutic applications. Anal. Bioanal. Chem. 2013, 405, 9149–9157. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukarasu, D.; Chen, T.; Liu, Z.; Hongdilokkul, N.; Romesberg, F.E. Selection of 2′-fluoro-modified aptamers with optimized properties. J. Am. Chem. Soc. 2017, 139, 2892–2895. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.S.; Hansen, L.H.; Vester, B.; Wengel, J. Improvement of a streptavidin-binding aptamer by LNA-and α-l-LNA-substitutions. Bioorg. Med. Chem. Lett. 2014, 24, 2273–2277. [Google Scholar] [CrossRef]
- Pasternak, A.; Hernandez, F.J.; Rasmussen, L.M.; Vester, B.; Wengel, J. Improved thrombin binding aptamer by incorporation of a single unlocked nucleic acid monomer. Nucleic Acids Res. 2011, 39, 1155–1164. [Google Scholar] [CrossRef]
- Cai, B.; Yang, X.; Sun, L.; Fan, X.; Li, L.; Jin, H.; Wu, Y.; Guan, Z.; Zhang, L.; Zhang, L. Stability and bioactivity of thrombin binding aptamers modified with D-/L-isothymidine in the loop regions. Org. Biomol. Chem. 2014, 12, 8866–8876. [Google Scholar] [CrossRef]
- Dalgaard, L.T.; Eliasson, L. An ‘alpha-beta’of pancreatic islet microribonucleotides. Int. J. Biochem. Cell Bio. 2017, 88, 208–219. [Google Scholar] [CrossRef]
- Li, L.; Yang, X.; Li, K.; Zhang, G.; Ma, Y.; Cai, B.; Li, S.; Ding, H.; Deng, J.; Nan, X. d-/l-Isothymidine incorporation in the core sequence of aptamer BC15 enhanced its binding affinity to the hnRNP A1 protein. Org. Biomol. Chem. 2018, 16, 7488–7497. [Google Scholar] [CrossRef]
- Li, K.; Deng, J.; Jin, H.; Yang, X.; Fan, X.; Li, L.; Zhao, Y.; Guan, Z.; Wu, Y.; Zhang, L. Chemical modification improves the stability of the DNA aptamer GBI-10 and its affinity towards tenascin-C. Org. Biomol. Chem. 2017, 15, 1174–1182. [Google Scholar] [CrossRef]
- Fan, X.; Sun, L.; Wu, Y.; Zhang, L.; Yang, Z. Bioactivity of 2′-deoxyinosine-incorporated aptamer AS1411. Sci. Rep. 2016, 6, 25799. [Google Scholar]
- Kratschmer, C.; Levy, M. Effect of chemical modifications on aptamer stability in serum. Nucleic Acid Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Narayan, C.; Veeramani, S.; Thiel, W.H. Optimization of RNA aptamer SELEX methods: Improved aptamer transcript 3′-end homogeneity, PAGE purification yield, and target-bound aptamer RNA recovery. Nucleic Acid Ther. 2022, 32, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zhang, Z.; Wang, B.; Chen, G.; Zhang, Y.; Deng, H.; Tang, Z.; Mao, J.; Wang, L. Combination Chemotherapy of Lung Cancer-Co-Delivery of Docetaxel Prodrug and Cisplatin Using Aptamer-Decorated Lipid-Polymer Hybrid Nanoparticles. Drug Des. Devel. Ther. 2020, 14, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, M.; Oskuee, R.K.; Nassirli, H.; Malaekeh-Nikouei, B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Control Release 2016, 236, 1–14. [Google Scholar] [CrossRef]
- He, S.; Du, Y.; Tao, H.; Duan, H. Advances in aptamer-mediated targeted delivery system for cancer treatment. Int. J. Biol. Macromol. 2023, 238, 124173. [Google Scholar] [CrossRef]
- Zhang, H.L.; Wang, L. N-acetylgalactosamine delivery systems for RNA therapeutics: A patent perspective. Expert Opin. Ther. Pat. 2023, 33, 539–547. [Google Scholar] [CrossRef]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Qiu, L.; Jing, Q.; Li, Y.; Han, J. RNA modification: Mechanisms and therapeutic targets. Mol. Biomed. 2023, 4, 25. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Ibba, M.L.; Ciccone, G.; Esposito, C.L.; Catuogno, S.; Giangrande, P.H. Advances in mRNA non-viral delivery approaches. Adv. Drug Deliv. Rev. 2021, 177, 113930. [Google Scholar] [CrossRef] [PubMed]
- Riddihough, G. Spotting Invaders. Sci. STKE 2006, 361, tw391. [Google Scholar] [CrossRef]
- Zhao, S.; Gao, K.; Han, H.; Stenzel, M.; Yin, B.; Song, H.; Lawanprasert, A.; Nielsen, J.E.; Sharma, R.; Arogundade, O.H.; et al. Acid-degradable lipid nanoparticles enhance the delivery of mRNA. Nat. Nanotechnol. 2024, 19, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef]
- Cao, Q.; Fang, H.; Tian, H. mRNA vaccines contribute to innate and adaptive immunity to enhance immune response in vivo. Biomaterials 2024, 310, 122628. [Google Scholar] [CrossRef]
- Janik, E.; Niemcewicz, M.; Ceremuga, M.; Krzowski, L.; Saluk-Bijak, J.; Bijak, M. Various aspects of a gene editing system—Crispr–cas9. Int. J. Mol. Sci. 2020, 21, 9604. [Google Scholar] [CrossRef]
- Li, J.; Shou, J.; Wu, Q. DNA fragment editing of genomes by CRISPR/Cas9. Yi Chuan 2015, 37, 992–1002. [Google Scholar]
- Hoy, S.M. Lesinurad: First Global Approval. Drugs 2016, 76, 509–516. [Google Scholar] [CrossRef]
- Prajapati, A.; Nain, V.; Singh, D. Designed gRNAs for CRISPR-Cas9 based antifungal resistance in eggplant. Bioinformation 2023, 19, 844. [Google Scholar]
- Tao, R.; Han, X.; Bai, X.; Yu, J.; Ma, Y.; Chen, W.; Zhang, D.; Li, Z. Revolutionizing cancer treatment: Enhancing CAR-T cell therapy with CRISPR/Cas9 gene editing technology. Front. Immunol. 2024, 15, 1354825. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhattacharjee, O.; Mandal, D.; Sen, M.K.; Dey, D.; Dasgupta, A.; Kazi, T.A.; Gupta, R.; Sinharoy, S.; Acharya, K.; et al. CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 2019, 232, 116636. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.X.; Fu, Y.W.; Zhao, J.J.; Zhang, F.; Li, S.A.; Zhao, M.; Wen, W.; Zhang, L.; Cheng, T.; Zhang, J.P.; et al. Superior Fidelity and Distinct Editing Outcomes of SaCas9 Compared with SpCas9 in Genome Editing. Genom. Proteom. Bioinform. 2023, 21, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Hu, L.; Shen, T.; Yang, R.; Jiang, L. Recent Advances in Gene Therapy for Familial Hypercholesterolemia: An Update Review. J. Clin. Med. 2022, 11, 6773. [Google Scholar] [CrossRef]
- Lee, J.; Lim, K.; Kim, A.; Mok, Y.G.; Chung, E.; Cho, S.I.; Lee, J.M.; Kim, J.S. Prime editing with genuine Cas9 nickases minimizes unwanted indels. Nat. Commun. 2023, 14, 1786. [Google Scholar] [CrossRef]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef]
- Liu, J.; Chang, J.; Jiang, Y.; Meng, X.; Sun, T.; Mao, L.; Xu, Q.; Wang, M. Fast and Efficient CRISPR/Cas9 Genome Editing In Vivo Enabled by Bioreducible Lipid and Messenger RNA Nanoparticles. Adv. Mater. 2019, 31, e1902575. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Coelho, T.; Marques, W., Jr.; Dasgupta, N.R.; Chao, C.-C.; Parman, Y.; França, M.C., Jr.; Guo, Y.-C.; Wixner, J.; Ro, L.-S.; Calandra, C.R.; et al. Eplontersen for hereditary transthyretin amyloidosis with polyneuropathy. JAMA 2023, 330, 1448–1458. [Google Scholar] [CrossRef]
- Wagner, K.R.; Kuntz, N.L.; Koenig, E.; East, L.; Upadhyay, S.; Han, B.; Shieh, P.B. Safety, tolerability, and pharmacokinetics of casimersen in patients with duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 2021, 64, 285–292. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First global approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Wilton-Clark, H.; Yokota, T. Recent trends in antisense therapies for Duchenne muscular dystrophy. Pharmaceutics 2023, 15, 778. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, O.; Yokota, T. Pharmacology and toxicology of eteplirsen and SRP-5051 for DMD exon 51 skipping: An update. Arch. Toxicol. 2022, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M. Genetics of amyotrophic lateral sclerosis: Seeking therapeutic targets in the era of gene therapy. J. Hum. Genet. 2023, 68, 131–152. [Google Scholar] [CrossRef]
- Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene therapy in amyotrophic lateral sclerosis. Cells 2022, 11, 2066. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Mccartan, R.; Khorkova, O.; Volmar, C.H.; Wahlestedt, C. Nucleic acid-based therapeutics for the treatment of central nervous system disorders. Front. Genet. 2023, 14, 1250276. [Google Scholar] [CrossRef]
- Scott, L.J.; Keam, S.J. Lumasiran: First approval. Drugs 2021, 81, 277–282. [Google Scholar] [CrossRef]
- Gulley, J.L.; Schlom, J.; Barcellos-Hoff, M.H.; Wang, X.-J.; Seoane, J.; Audhuy, F.; Lan, Y.; Dussault, I.; Moustakas, A. Dual inhibition of TGF-beta and PD-L1: A novel approach to cancer treatment. Mol. Oncol. 2022, 16, 2117–2134. [Google Scholar] [CrossRef]
- Andrews, D.W.; Judy, K.D.; Scott, C.B.; Garcia, S.; Harshyne, L.A.; Kenyon, L.; Talekar, K.; Flanders, A.; Atsina, K.-B.; Kim, L.; et al. Phase Ib clinical trial of IGV-001 for patients with newly diagnosed glioblastoma. Clin. Cancer Res. 2021, 27, 1912–1922. [Google Scholar]
- Yao, R.; Xie, C.; Xia, X. Recent progress in mRNA cancer vaccines. Hum. Vaccines Immun. 2024, 20, 2307187. [Google Scholar]
- Nakamura, Y. Multiple therapeutic applications of RBM-007, an anti-FGF2 aptamer. Cells 2021, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.; Ruz, V.; Rico, L.; Martinez, T.; Monteiro, S.; Cuesta, A.; Guerra, A.; Cuenca, A.; Gonzalez, V. SYL1801: Preclinical efficacy and safety of a sirna-based eye drops treatment for age related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5389. [Google Scholar]
- Moreno-Montanes, J.; Bleau, A.M.; Jimenez, A.I. Tivanisiran, a novel siRNA for the treatment of dry eye disease. Expert Opin. Investig. Drugs 2018, 27, 421–426. [Google Scholar] [CrossRef]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; van Wijk, E.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.J.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Lawrence, D.; Friedman, A.; et al. Inclisiran and cardiovascular events: A patient-level analysis of phase III trials. Eur. Heart J. 2023, 44, 129–138. [Google Scholar] [CrossRef]
- Szabo, G.T.; Mahiny, A.J.; Vlatkovic, I. COVID-19 mRNA vaccines: Platforms and current developments. Mol. Ther. 2022, 30, 1850–1868. [Google Scholar] [CrossRef]
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target Ther. 2022, 7, 94. [Google Scholar] [CrossRef]
- Baden, L.R.; Sahly, H.M.E.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, B.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Yuen, M.F.; Heo, J.; Jang, J.-W.; Yoon, J.-H.; Kweon, Y.-O.; Park, S.-J.; Tami, Y.; You, S.; Yates, P.; Tao, Y.; et al. Safety, tolerability and antiviral activity of the antisense oligonucleotide bepirovirsen in patients with chronic hepatitis B: A phase 2 randomized controlled trial. Nat. Med. 2021, 27, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Asselah, T.; Jacobson, P.I.M.; Brunetto, M.R.; Janssen, H.L.A.; Takehara, T.; Hou, J.L.; Kakuda, T.N.; Lambrecht, T.; Beumont, M.; et al. Efficacy and safety of the siRNA JNJ-73763989 and the capsid assembly modulator JNJ-56136379 (bersacapavir) with nucleos(t)ide analogues for the treatment of chronic hepatitis B virus infection (REEF-1): A multicentre, double-blind, active-controlled, randomised, phase 2b trial. Lancet Gastroenterol. Hepatol. 2023, 8, 790–802. [Google Scholar] [PubMed]
- Gupta, S.V.; Fanget, M.C.; MacLauchlin, C.; Clausen, V.A.; Li, J.; Cloutier, D.; Shen, L.; Robbie, G.J.; Mogalian, E. Clinical and preclinical single-dose pharmacokinetics of VIR-2218, an RNAi therapeutic targeting HBV infection. Drugs R D 2021, 21, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V. Hepatitis B gene therapy coming to age. AIDS Rev. 2018, 20, 125–127. [Google Scholar]
- Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888. [Google Scholar] [CrossRef]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjugate Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef]
- Gao, X.; Yao, L.; Song, Q.; Zhu, L.; Xia, Z.; Xia, H.; Jiang, X.; Chen, J.; Chen, H. The association of autophagy with polyethylenimine-induced cytotoxicity in nephritic and hepatic cell lines. Biomaterials 2011, 32, 8613–8625. [Google Scholar] [CrossRef]
- Hashida, M. Role of pharmacokinetic consideration for the development of drug delivery systems: A historical overview. Adv. Drug Deliv. Rev. 2020, 157, 71–82. [Google Scholar] [CrossRef]
- Jiang, R.; Shirin, H.; Marsha, R.E.; Cassandra, Y.; Estevan, S.Z.; Raku, S. Factors influencing ADME properties of therapeutic antisense oligonucleotides: Physicochemical characteristics and beyond. Curr. Drug Metab. 2023, 24, 536–552. [Google Scholar] [CrossRef]
- Alhamadani, F.; Zhang, K.; Parikh, R.; Wu, H.; Rasmussen, T.P.; Bahal, R.; Zhong, X.-B.; Manautou, J.E. Adverse drug reactions and toxicity of the food and drug administration-approved antisense oligonucleotide drugs. Drug Metab. Dispos. 2022, 50, 879–887. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wang, C.; Fu, X.; Ren, M. The Progress and Evolving Trends in Nucleic-Acid-Based Therapies. Biomolecules 2025, 15, 376. https://doi.org/10.3390/biom15030376
Liu Y, Wang C, Fu X, Ren M. The Progress and Evolving Trends in Nucleic-Acid-Based Therapies. Biomolecules. 2025; 15(3):376. https://doi.org/10.3390/biom15030376
Chicago/Turabian StyleLiu, Yunlong, Chunmiao Wang, Xiuping Fu, and Mengtian Ren. 2025. "The Progress and Evolving Trends in Nucleic-Acid-Based Therapies" Biomolecules 15, no. 3: 376. https://doi.org/10.3390/biom15030376
APA StyleLiu, Y., Wang, C., Fu, X., & Ren, M. (2025). The Progress and Evolving Trends in Nucleic-Acid-Based Therapies. Biomolecules, 15(3), 376. https://doi.org/10.3390/biom15030376