
The Regulation of Cellular Senescence in Cancer

 , ,
, , 
Abstract
:1. Introduction
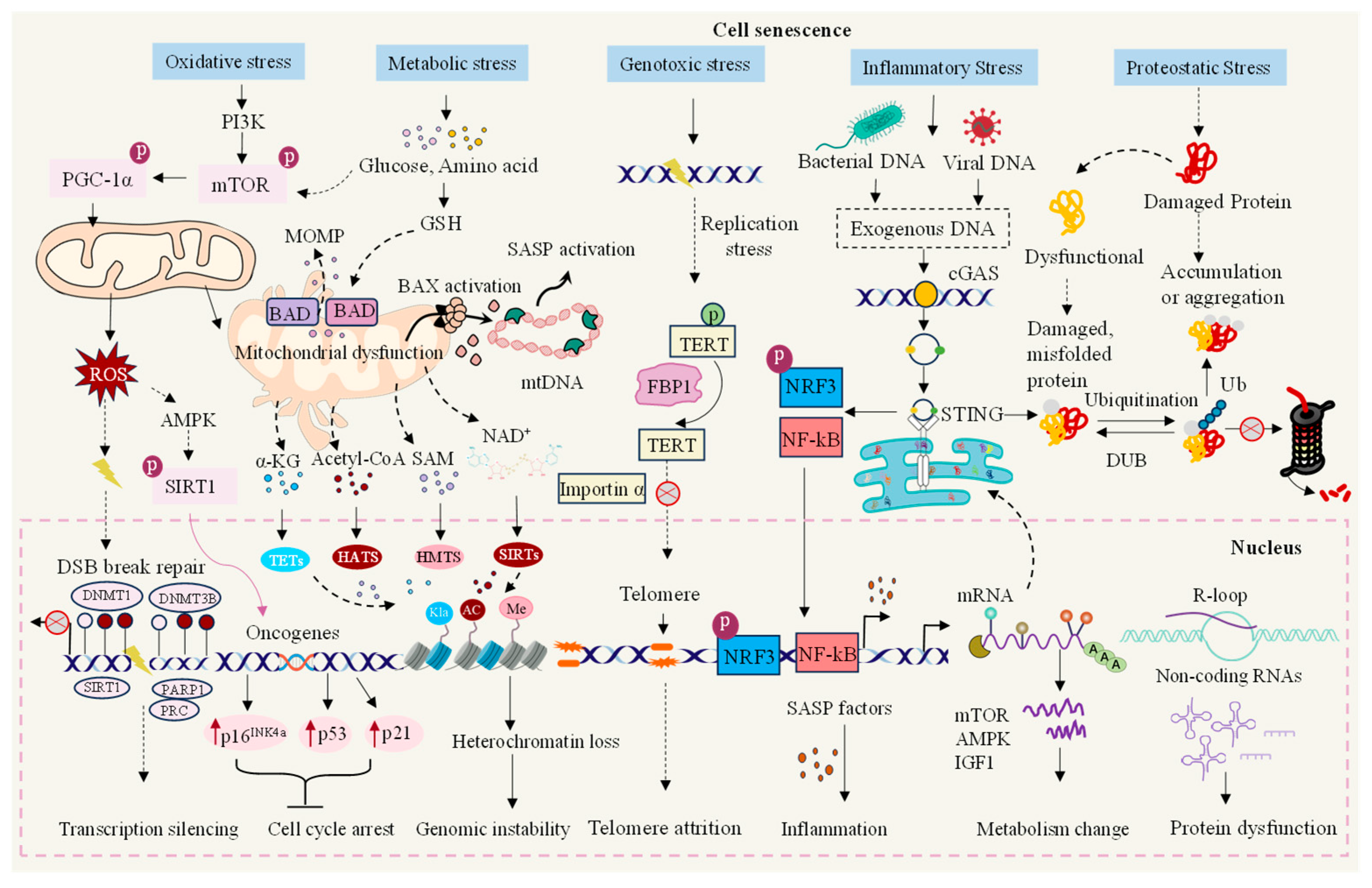
2. The Causes of Normal Cellular Senescence
2.1. Telomere Shortening
2.2. Genomic Instability and Epigenetic Change
2.3. Metabolic Changes
2.4. Abnormalities in Signal Transduction
2.4.1. AMPK/mTORC Signaling Pathway
2.4.2. Insulin-like Signaling Pathways
2.4.3. Sirtuin-Mediated Signaling Pathway
3. Senescence of Immune Cells
3.1. Innate Immunity in Senescence
3.2. Adaptive Immunity in Senescence
4. The Causes of Cancer Cellular Senescence
4.1. Oncogene Gene-Induced Senescence
4.2. Drug-Induced Senescence
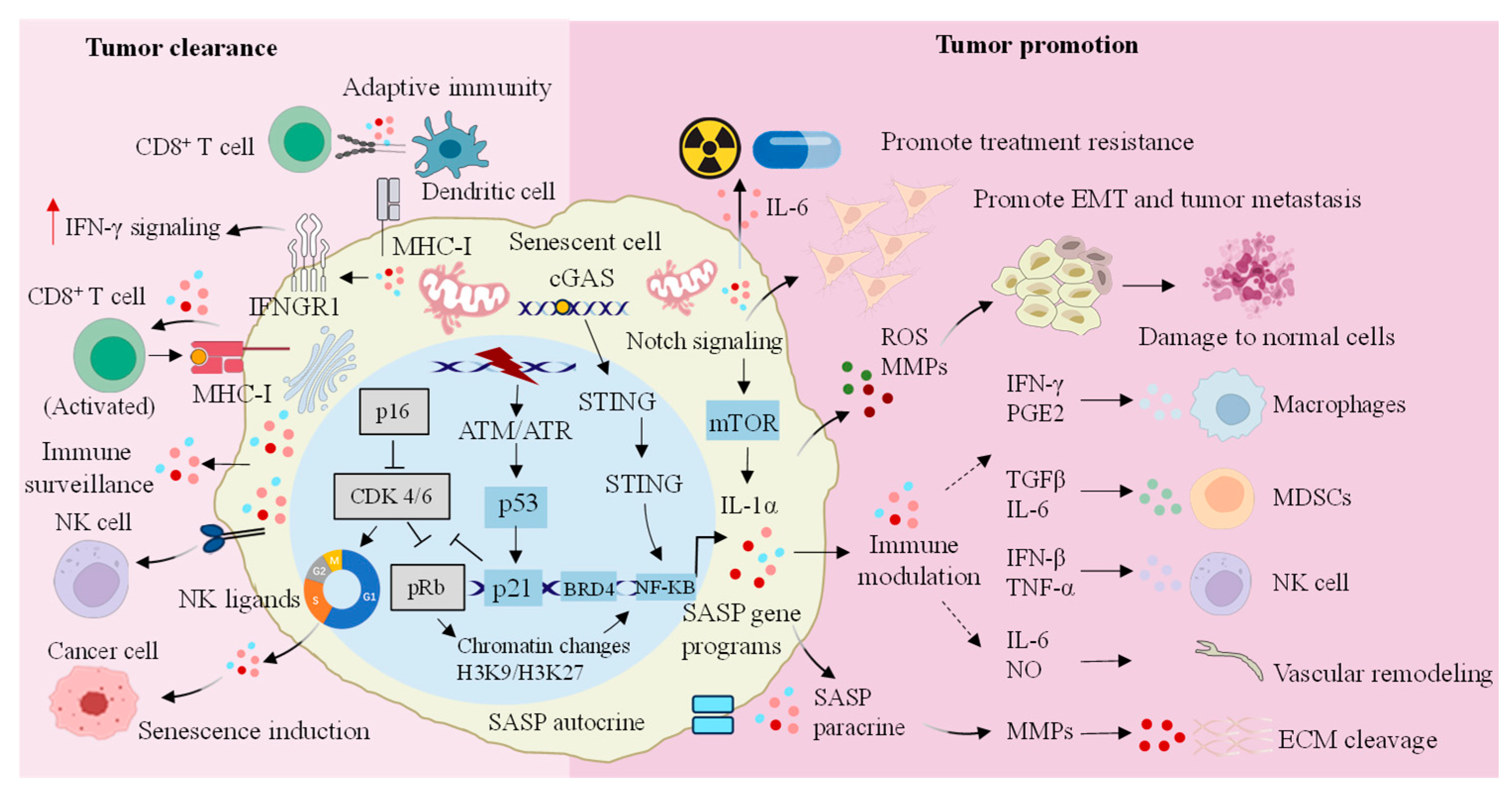
5. The Paradoxical Role of Cellular Senescence in Cancer
5.1. Senescent Cells Suppress Tumors
5.1.1. Growth Retardation
5.1.2. Immune Surveillance
5.2. Senescence and Tumor Promotion
5.2.1. Senescence-Mediated Remodeling of Tumor Microenvironment
5.2.2. Immune Escape Mediated by Cell Senescence
6. Senescence Therapy of Tumor Cells
7. Discussion and Prospect
Author Contributions
Funding
Conflicts of Interest
References
- Mullard, A. Addressing cancer’s grand challenges. Nat. Rev. Drug Discov. 2020, 19, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.; Rahrmann, E.P.; Simons, B.D.; Gilbertson, R.J. Cancers make their own luck: Theories of cancer origins. Nat. Rev. Cancer 2023, 23, 710–724. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Prieto, L.I.; Sturmlechner, I.; Goronzy, J.J.; Baker, D.J. Senescent cells as thermostats of antitumor immunity. Sci. Transl. Med. 2023, 15, eadg7291. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar]
- Liu, X.-L.; Ding, J.; Meng, L.-H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- López-Otín, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 12–35. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—Role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes. Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Prieto, L.I.; Baker, D.J. Cellular Senescence and the Immune System in Cancer. Gerontology 2019, 65, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.; Tao, J.; Jiang, H.; Yang, H.; Guo, D.; Zhao, H.; He, X.; Luo, S.; Jiang, X.; et al. Fructose-1,6-bisphosphatase 1 dephosphorylates and inhibits TERT for tumor suppression. Nat. Chem. Biol. 2024, 20, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef]
- Lanna, A.; Vaz, B.; D’Ambra, C.; Valvo, S.; Vuotto, C.; Chiurchiù, V.; Devine, O.; Sanchez, M.; Borsellino, G.; Akbar, A.N.; et al. An intercellular transfer of telomeres rescues T cells from senescence and promotes long-term immunological memory. Nat. Cell Biol. 2022, 24, 1461–1474. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Vandiver, A.R.; Hoang, A.N.; Herbst, A.; Lee, C.C.; Aiken, J.M.; McKenzie, D.; Teitell, M.A.; Timp, W.; Wanagat, J. Nanopore sequencing identifies a higher frequency and expanded spectrum of mitochondrial DNA deletion mutations in human aging. Aging Cell 2023, 22, e13842. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef]
- Yang, L.; Ruan, Z.; Lin, X.; Wang, H.; Xin, Y.; Tang, H.; Hu, Z.; Zhou, Y.; Wu, Y.; Wang, J.; et al. NAD+ dependent UPRmt activation underlies intestinal aging caused by mitochondrial DNA mutations. Nat. Commun. 2024, 15, 546. [Google Scholar] [CrossRef] [PubMed]
- Seale, K.; Horvath, S.; Teschendorff, A.; Eynon, N.; Voisin, S. Making sense of the ageing methylome. Nat Rev Genet. 2022, 23, 585–605. [Google Scholar] [CrossRef]
- Yang, J.-H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e27. [Google Scholar] [CrossRef]
- Yao, J.; Ding, D.; Li, X.; Shen, T.; Fu, H.; Zhong, H.; Wei, G.; Ni, T. Prevalent intron retention fine-tunes gene expression and contributes to cellular senescence. Aging Cell 2020, 19, e13276. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Lyu, G.; Han, M.; Nie, H.; Shen, T.; Chen, W.; Niu, Y.; Song, Y.; Li, X.; Li, H.; et al. 3′ UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Res. 2018, 28, 285–294. [Google Scholar] [CrossRef]
- Mortuza, R.; Chen, S.; Feng, B.; Sen, S.; Chakrabarti, S. High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS ONE 2013, 8, e54514. [Google Scholar] [CrossRef]
- Dou, X.; Fu, Q.; Long, Q.; Liu, S.; Zou, Y.; Fu, D.; Xu, Q.; Jiang, Z.; Ren, X.; Zhang, G.; et al. PDK4-dependent hypercatabolism and lactate production of senescent cells promotes cancer malignancy. Nat. Metab. 2023, 5, 1887–1910. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, L.; Huang, H.; Yu, Q.; Hu, B.; Wang, G.; Ge, F.; Yin, T.; Li, S.; Yu, X. Phosphoglycerate dehydrogenase activates PKM2 to phosphorylate histone H3T11 and attenuate cellular senescence. Nat. Commun. 2023, 14, 1323. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hartman, C.L.; Li, L.; Albert, C.J.; Si, F.; Gao, A.; Huang, L.; Zhao, Y.; Lin, W.; Hsueh, E.C.; et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci. Transl. Med. 2021, 13, eaaz6314. [Google Scholar] [CrossRef]
- Ripa, R.; Ballhysa, E.; Steiner, J.D.; Laboy, R.; Annibal, A.; Hochhard, N.; Latza, C.; Dolfi, L.; Calabrese, C.; Meyer, A.M.; et al. Refeeding-associated AMPKγ1 complex activity is a hallmark of health and longevity. Nat. Aging 2023, 3, 1544–1560. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, S.; Zhuang, Y.; Xie, F.; Wang, R.; Kong, X.; Zhang, Q.; Feng, Y.; Gao, H.; Kong, X.; et al. Muscle PARP1 inhibition extends lifespan through AMPKα PARylation and activation in Drosophila. Proc. Natl. Acad. Sci. USA 2023, 120, e2213857120. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Syntichaki, P.; Troulinaki, K.; Tavernarakis, N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 2007, 445, 922–926. [Google Scholar] [CrossRef]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar]
- Arif, A.; Terenzi, F.; Potdar, A.A.; Jia, J.; Sacks, J.; China, A.; Halawani, D.; Vasu, K.; Li, X.; Brown, J.M.; et al. EPRS is a critical mTORC1-S6K1 effector that influences adiposity in mice. Nature 2017, 542, 357–361. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef]
- Powolny, A.A.; Singh, S.V.; Melov, S.; Hubbard, A.; Fisher, A.L. The garlic constituent diallyl trisulfide increases the lifespan of C. elegans via skn-1 activation. Exp. Gerontol. 2011, 46, 441–452. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.O.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef]
- Sirozh, O.; Saez-Mas, A.; Jung, B.; Sanchez-Burgos, L.; Zarzuela, E.; Rodrigo-Perez, S.; Ventoso, I.; Lafarga, V.; Fernandez-Capetillo, O. Nucleolar stress caused by arginine-rich peptides triggers a ribosomopathy and accelerates aging in mice. Mol. Cell 2024, 84, 1527–1540.e7. [Google Scholar] [CrossRef]
- Ortega-Molina, A.; Lebrero-Fernández, C.; Sanz, A.; Calvo-Rubio, M.; Deleyto-Seldas, N.; de Prado-Rivas, L.; Plata-Gómez, A.B.; Fernández-Florido, E.; González-García, P.; Vivas-García, Y.; et al. A mild increase in nutrient signaling to mTORC1 in mice leads to parenchymal damage, myeloid inflammation and shortened lifespan. Nat. Aging 2024, 4, 1102–1120. [Google Scholar] [CrossRef]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Tyshkovskiy, A.; Ma, S.; Shindyapina, A.V.; Tikhonov, S.; Lee, S.-G.; Bozaykut, P.; Castro, J.P.; Seluanov, A.; Schork, N.J.; Gorbunova, V.; et al. Distinct longevity mechanisms across and within species and their association with aging. Cell 2023, 186, 2929–2949.e20. [Google Scholar] [CrossRef]
- Baghdadi, M.; Nespital, T.; Mesaros, A.; Buschbaum, S.; Withers, D.J.; Grönke, S.; Partridge, L. Reduced insulin signaling in neurons induces sex-specific health benefits. Sci. Adv. 2023, 9, eade8137. [Google Scholar] [CrossRef]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef]
- Lin, S.J.; Defossez, P.A.; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef]
- Ye, Y.; Yang, K.; Liu, H.; Yu, Y.; Song, M.; Huang, D.; Lei, J.; Zhang, Y.; Liu, Z.; Chu, Q.; et al. SIRT2 counteracts primate cardiac aging via deacetylation of STAT3 that silences CDKN2B. Nat. Aging 2023, 3, 1269–1287. [Google Scholar]
- Zhang, X.; Liu, T.; Hou, X.; Zhou, Z.; Zhang, F.; Ma, H.; Wu, X.; Jiang, J. Exosomes secreted by mesenchymal stem cells delay brain aging by upregulating SIRT1 expression. Sci. Rep. 2023, 13, 13213. [Google Scholar] [CrossRef]
- Bi, S.; Jiang, X.; Ji, Q.; Wang, Z.; Ren, J.; Wang, S.; Yu, Y.; Wang, R.; Liu, Z.; Liu, J.; et al. The sirtuin-associated human senescence program converges on the activation of placenta-specific gene PAPPA. Dev. Cell 2024, 59, 991–1009.e12. [Google Scholar] [CrossRef]
- Yang, F.; Deng, X.; Yu, Y.; Luo, L.; Chen, X.; Zheng, J.; Qiu, Y.; Xiao, F.; Xie, X.; Zhao, Y.; et al. Association of Human Whole Blood NAD+ Contents with Aging. Front. Endocrinol. 2022, 13, 829658. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Gabrawy, M.M.; Westbrook, R.; King, A.; Khosravian, N.; Ochaney, N.; DeCarvalho, T.; Wang, Q.; Yu, Y.; Huang, Q.; Said, A.; et al. Dual treatment with kynurenine pathway inhibitors and NAD+ precursors synergistically extends life span in Drosophila. Aging Cell 2024, 23, e14102. [Google Scholar] [CrossRef]
- Walford, R.L. The immunologic theory of aging. Gerontologist 1964, 4, 195–197. [Google Scholar] [CrossRef]
- Reed, R.G. Stress and Immunological Aging. Curr. Opin. Behav. Sci. 2019, 28, 38–43. [Google Scholar] [CrossRef]
- Liu, Z.; Liang, Q.; Ren, Y.; Guo, C.; Ge, X.; Wang, L.; Cheng, Q.; Luo, P.; Zhang, Y.; Han, X. Immunosenescence: Molecular mechanisms and diseases. Signal Transduct. Target. Ther. 2023, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.-A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Luo, O.J.; Lei, W.; Zhu, G.; Ren, Z.; Xu, Y.; Xiao, C.; Zhang, H.; Cai, J.; Luo, Z.; Gao, L.; et al. Multidimensional single-cell analysis of human peripheral blood reveals characteristic features of the immune system landscape in aging and frailty. Nat. Aging 2022, 2, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res. Rev. 2013, 12, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Hampson, P.; Lord, J.M. Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell 2012, 11, 751–759. [Google Scholar] [CrossRef]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Van Vré, E.A.; Van Brussel, I.; Bosmans, J.M.; Vrints, C.J.; Bult, H. Dendritic cells in human atherosclerosis: From circulation to atherosclerotic plaques. Mediat. Inflamm. 2011, 2011, 941396. [Google Scholar] [CrossRef]
- Agrawal, A.; Gupta, S. Impact of aging on dendritic cell functions in humans. Ageing Res. Rev. 2011, 10, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Chougnet, C.A.; Thacker, R.I.; Shehata, H.M.; Hennies, C.M.; Lehn, M.A.; Lages, C.S.; Janssen, E.M. Loss of Phagocytic and Antigen Cross-Presenting Capacity in Aging Dendritic Cells Is Associated with Mitochondrial Dysfunction. J. Immunol. 2015, 195, 2624–2632. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.J.; Lalinde Ruiz, N.; Llano León, M.; Martínez Enríquez, L.; Montilla Velásquez, M.D.P.; Ortiz Aguirre, J.P.; Rodríguez Bohórquez, O.M.; Velandia Vargas, E.A.; Hernández, E.D.; Parra López, C.A. Immunosenescence Study of T Cells: A Systematic Review. Front. Immunol. 2020, 11, 604591. [Google Scholar] [CrossRef] [PubMed]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef]
- Baggiolini, A.; Callahan, S.J.; Montal, E.; Weiss, J.M.; Trieu, T.; Tagore, M.M.; Tischfield, S.E.; Walsh, R.M.; Suresh, S.; Fan, Y.; et al. Developmental chromatin programs determine oncogenic competence in melanoma. Science 2021, 373, eabc1048. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Wang, H.; Lu, J.; Stevens, T.; Roberts, A.; Mandel, J.; Avula, R.; Ma, B.; Wu, Y.; Wang, J.; Land, C.V.; et al. Premature aging and reduced cancer incidence associated with near-complete body-wide Myc inactivation. Cell Rep. 2023, 42, 112830. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Litwiniec, A.; Gackowska, L.; Helmin-Basa, A.; Zuryń, A.; Grzanka, A. Low-dose etoposide-treatment induces endoreplication and cell death accompanied by cytoskeletal alterations in A549 cells: Does the response involve senescence? The possible role of vimentin. Cancer Cell Int. 2013, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Fu, V.X.; Desotelle, J.A.; Kenowski, M.L.; Jarrard, D.F. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia 2005, 7, 816–823. [Google Scholar]
- Ewald, J.A.; Peters, N.; Desotelle, J.A.; Hoffmann, F.M.; Jarrard, D.F. A high-throughput method to identify novel senescence-inducing compounds. J. Biomol. Screen. 2009, 14, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Gaumont-Leclerc, M.-F.; Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes. Dev. 2007, 21, 43–48. [Google Scholar]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef]
- Dabrowska, M.; Skoneczny, M.; Uram, L.; Rode, W. Methotrexate-induced senescence of human colon cancer cells depends on p53 acetylation, but not genomic aberrations. Anticancer. Drugs 2019, 30, 374–382. [Google Scholar] [CrossRef]
- Song, Y.; Baba, T.; Mukaida, N. Gemcitabine induces cell senescence in human pancreatic cancer cell lines. Biochem. Biophys. Res. Commun. 2016, 477, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence—Its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Kumarasamy, V.; Nambiar, R.; Pearson, J.D.; Vail, P.; Rosenheck, H.; Wang, J.; Eng, K.; Bremner, R.; Schramek, D.; et al. CDK/cyclin dependencies define extreme cancer cell-cycle heterogeneity and collateral vulnerabilities. Cell Rep. 2022, 38, 110448. [Google Scholar] [CrossRef] [PubMed]
- Crozier, L.; Foy, R.; Adib, R.; Kar, A.; Holt, J.A.; Pareri, A.U.; Valverde, J.M.; Rivera, R.; Weston, W.A.; Wilson, R.; et al. CDK4/6 inhibitor-mediated cell overgrowth triggers osmotic and replication stress to promote senescence. Mol. Cell 2023, 83, 4062–4077.e5. [Google Scholar] [CrossRef]
- Dietrich, C.; Trub, A.; Ahn, A.; Taylor, M.; Ambani, K.; Chan, K.T.; Lu, K.-H.; Mahendra, C.A.; Blyth, C.; Coulson, R.; et al. INX-315, a Selective CDK2 Inhibitor, Induces Cell Cycle Arrest and Senescence in Solid Tumors. Cancer Discov. 2024, 14, 446–467. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gomez, D.L.M.; Armando, R.G.; Cerrudo, C.S.; Ghiringhelli, P.D.; Gomez, D.E. Telomerase as a Cancer Target. Development of New Molecules. Curr. Top. Med. Chem. 2016, 16, 2432–2440. [Google Scholar]
- Yu, S.; Wei, S.; Savani, M.; Lin, X.; Du, K.; Mender, I.; Siteni, S.; Vasilopoulos, T.; Reitman, Z.J.; Ku, Y.; et al. A Modified Nucleoside 6-Thio-2′-Deoxyguanosine Exhibits Antitumor Activity in Gliomas. Clin. Cancer Res. 2021, 27, 6800–6814. [Google Scholar] [CrossRef]
- Liang, W.-W.; Lu, R.J.-H.; Jayasinghe, R.G.; Foltz, S.M.; Porta-Pardo, E.; Geffen, Y.; Wendl, M.C.; Lazcano, R.; Kolodziejczak, I.; Song, Y.; et al. Integrative multi-omic cancer profiling reveals DNA methylation patterns associated with therapeutic vulnerability and cell-of-origin. Cancer Cell 2023, 41, 1567–1585.e7. [Google Scholar] [CrossRef]
- Amatori, S.; Bagaloni, I.; Viti, D.; Fanelli, M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 2011, 71, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, A.; Wei, Y.; Liu, F.; Li, P.; Fang, R.; Ma, L.; Zhang, S.; Wang, L.; Liu, J.; et al. Critical role of lncEPAT in coupling dysregulated EGFR pathway and histone H2A deubiquitination during glioblastoma tumorigenesis. Sci. Adv. 2022, 8, eabn2571. [Google Scholar] [CrossRef]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease: Emerging Therapeutic Strategies. JAMA 2018, 320, 1319–1320. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Cao, S.; Xu, R. Cancer incidence, mortality, and burden in China: A time-trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun. 2021, 41, 1037–1048. [Google Scholar] [CrossRef]
- Kadambi, S.; Loh, K.P.; Dunne, R.; Magnuson, A.; Maggiore, R.; Zittel, J.; Flannery, M.; Inglis, J.; Gilmore, N.; Mohamed, M.; et al. Older adults with cancer and their caregivers—Current landscape and future directions for clinical care. Nat. Rev. Clin. Oncol. 2020, 17, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef]
- Wilson, G.A.; Vuina, K.; Sava, G.; Huard, C.; Meneguello, L.; Coulombe-Huntington, J.; Bertomeu, T.; Maizels, R.J.; Lauring, J.; Kriston-Vizi, J.; et al. Active growth signaling promotes senescence and cancer cell sensitivity to CDK7 inhibition. Mol. Cell 2023, 83, 4078–4092.e6. [Google Scholar] [CrossRef]
- Innes, A.J.; Sun, B.; Wagner, V.; Brookes, S.; McHugh, D.; Pombo, J.; Porreca, R.M.; Dharmalingam, G.; Vernia, S.; Zuber, J.; et al. XPO7 is a tumor suppressor regulating p21CIP1-dependent senescence. Genes. Dev. 2021, 35, 379–391. [Google Scholar] [CrossRef]
- Nassour, J.; Aguiar, L.G.; Correia, A.; Schmidt, T.T.; Mainz, L.; Przetocka, S.; Haggblom, C.; Tadepalle, N.; Williams, A.; Shokhirev, M.N.; et al. Telomere-to-mitochondria signalling by ZBP1 mediates replicative crisis. Nature 2023, 614, 767–773. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Strittmatter, A.; Sunde, U.; Zegners, D. Life cycle patterns of cognitive performance over the long run. Proc. Natl. Acad. Sci. USA 2020, 117, 27255–27261. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.; Boix, O.; Garcia-Garijo, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.; Prats, N.; Lopez-Dominguez, J.A.; et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov. 2023, 13, 410–431. [Google Scholar] [CrossRef]
- Chen, H.-A.; Ho, Y.-J.; Mezzadra, R.; Adrover, J.M.; Smolkin, R.; Zhu, C.; Woess, K.; Bernstein, N.; Schmitt, G.; Fong, L.; et al. Senescence Rewires Microenvironment Sensing to Facilitate Antitumor Immunity. Cancer Discov. 2023, 13, 432–453. [Google Scholar] [CrossRef]
- Meng, Y.; Efimova, E.V.; Hamzeh, K.W.; Darga, T.E.; Mauceri, H.J.; Fu, Y.-X.; Kron, S.J.; Weichselbaum, R.R. Radiation-inducible immunotherapy for cancer: Senescent tumor cells as a cancer vaccine. Mol. Ther. 2012, 20, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- van Tuyn, J.; Jaber-Hijazi, F.; MacKenzie, D.; Cole, J.J.; Mann, E.; Pawlikowski, J.S.; Rai, T.S.; Nelson, D.M.; McBryan, T.; Ivanov, A.; et al. Oncogene-Expressing Senescent Melanocytes Up-Regulate MHC Class II, a Candidate Melanoma Suppressor Function. J. Investig. Dermatol. 2017, 137, 2197–2207. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Purohit, S.; Zhi, W.; Ferris, D.G.; Alverez, M.; Tran, L.K.H.; Tran, P.M.H.; Dun, B.; Hopkins, D.; Santos, B.D.; Ghamande, S.; et al. Senescence-Associated Secretory Phenotype Determines Survival and Therapeutic Response in Cervical Cancer. Cancers 2020, 12, 2899. [Google Scholar] [CrossRef]
- Liu, P.; Li, F.; Lin, J.; Fukumoto, T.; Nacarelli, T.; Hao, X.; Kossenkov, A.V.; Simon, M.C.; Zhang, R. m6A-independent genome-wide METTL3 and METTL14 redistribution drives the senescence-associated secretory phenotype. Nat. Cell Biol. 2021, 23, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Shahbandi, A.; Chiu, F.-Y.; Ungerleider, N.A.; Kvadas, R.; Mheidly, Z.; Sun, M.J.S.; Tian, D.; Waizman, D.A.; Anderson, A.Y.; Machado, H.L.; et al. Breast cancer cells survive chemotherapy by activating targetable immune-modulatory programs characterized by PD-L1 or CD80. Nat. Cancer 2022, 3, 1513–1533. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Fu, Y.; Stewart, S.A. Senescence and the pro-tumorigenic stroma. Crit. Rev. Oncog. 2013, 18, 549–558. [Google Scholar]
- Elkhattouti, A.; Hassan, M.; Gomez, C.R. Stromal Fibroblast in Age-Related Cancer: Role in Tumorigenesis and Potential as Novel Therapeutic Target. Front. Oncol. 2015, 5, 158. [Google Scholar] [CrossRef]
- Balliet, R.M.; Capparelli, C.; Guido, C.; Pestell, T.G.; Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Pestell, R.G.; Howell, A.; et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: Understanding the aging and cancer connection. Cell Cycle 2011, 10, 4065–4073. [Google Scholar] [CrossRef] [PubMed]
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The role of senescence in cancer development. Semin. Cancer Biol. 2020, 62, 182–191. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.-K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef]
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230. [Google Scholar] [CrossRef]
- Chen, F.; Long, Q.; Fu, D.; Zhu, D.; Ji, Y.; Han, L.; Zhang, B.; Xu, Q.; Liu, B.; Li, Y.; et al. Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat. Commun. 2018, 9, 4315. [Google Scholar] [CrossRef] [PubMed]
- Salam, R.; Saliou, A.; Bielle, F.; Bertrand, M.; Antoniewski, C.; Carpentier, C.; Alentorn, A.; Capelle, L.; Sanson, M.; Huillard, E.; et al. Cellular senescence in malignant cells promotes tumor progression in mouse and patient Glioblastoma. Nat. Commun. 2023, 14, 441. [Google Scholar] [CrossRef]
- Ji, J.; Ding, K.; Cheng, B.; Zhang, X.; Luo, T.; Huang, B.; Yu, H.; Chen, Y.; Xu, X.; Lin, H.; et al. Radiotherapy-Induced Astrocyte Senescence Promotes an Immunosuppressive Microenvironment in Glioblastoma to Facilitate Tumor Regrowth. Adv. Sci. 2024, 11, e2304609. [Google Scholar] [CrossRef]
- Li, W.; Kawaguchi, K.; Tanaka, S.; He, C.; Maeshima, Y.; Suzuki, E.; Toi, M. Cellular senescence triggers intracellular acidification and lysosomal pH alkalinized via ATP6AP2 attenuation in breast cancer cells. Commun. Biol. 2023, 6, 1147. [Google Scholar] [CrossRef]
- Wang, T.-W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef]
- Liu, X.; Li, L.; Si, F.; Huang, L.; Zhao, Y.; Zhang, C.; Hoft, D.F.; Peng, G. NK and NKT cells have distinct properties and functions in cancer. Oncogene 2021, 40, 4521–4537. [Google Scholar] [CrossRef]
- Colucci, M.; Zumerle, S.; Bressan, S.; Gianfanti, F.; Troiani, M.; Valdata, A.; D’Ambrosio, M.; Pasquini, E.; Varesi, A.; Cogo, F.; et al. Retinoic acid receptor activation reprograms senescence response and enhances anti-tumor activity of natural killer cells. Cancer Cell 2024, 42, 646–661.e9. [Google Scholar] [CrossRef] [PubMed]
- Vaena, S.; Chakraborty, P.; Lee, H.G.; Janneh, A.H.; Kassir, M.F.; Beeson, G.; Hedley, Z.; Yalcinkaya, A.; Sofi, M.H.; Li, H.; et al. Aging-dependent mitochondrial dysfunction mediated by ceramide signaling inhibits antitumor T cell response. Cell Rep. 2021, 35, 109076. [Google Scholar] [CrossRef]
- Haston, S.; Gonzalez-Gualda, E.; Morsli, S.; Ge, J.; Reen, V.; Calderwood, A.; Moutsopoulos, I.; Panousopoulos, L.; Deletic, P.; Carreno, G.; et al. Clearance of senescent macrophages ameliorates tumorigenesis in KRAS-driven lung cancer. Cancer Cell 2023, 41, 1242–1260.e6. [Google Scholar] [CrossRef]
- Prieto, L.I.; Sturmlechner, I.; Graves, S.I.; Zhang, C.; Goplen, N.P.; Yi, E.S.; Sun, J.; Li, H.; Baker, D.J. Senescent alveolar macrophages promote early-stage lung tumorigenesis. Cancer Cell 2023, 41, 1261–1275.e6. [Google Scholar] [CrossRef]
- Goldblatt, E.M.; Gentry, E.R.; Fox, M.J.; Gryaznov, S.M.; Shen, C.; Herbert, B.-S. The telomerase template antagonist GRN163L alters MDA-MB-231 breast cancer cell morphology, inhibits growth, and augments the effects of paclitaxel. Mol. Cancer Ther. 2009, 8, 2027–2035. [Google Scholar] [CrossRef]
- Chava, S.; Bugide, S.; Malvi, P.; Gupta, R. Co-targeting of specific epigenetic regulators in combination with CDC7 potently inhibit melanoma growth. iScience. 2022, 25, 104752. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.-C.; Ho, Y.-J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.-J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21. [Google Scholar] [CrossRef]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- van Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Chelyapov, N.; Nguyen, T.T.; Gonzalez, R. Autologous NK cells propagated and activated ex vivo decrease senescence markers in human PBMCs. Biochem. Biophys. Rep. 2022, 32, 101380. [Google Scholar] [CrossRef]
- Bai, Z.; Yang, P.; Yu, F.; Li, Z.; Yao, Z.; Martinez, J.; Li, M.; Xu, H. Combining adoptive NK cell infusion with a dopamine-releasing peptide reduces senescent cells in aged mice. Cell Death Dis. 2022, 13, 305. [Google Scholar] [CrossRef]
- Yuan, Y.; Li, H.; Pu, W.; Chen, L.; Guo, D.; Jiang, H.; He, B.; Qin, S.; Wang, K.; Li, N.; et al. Cancer metabolism and tumor microenvironment: Fostering each other? Sci. China Life Sci. 2022, 65, 236–279. [Google Scholar] [CrossRef] [PubMed]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Normal Cells | Cancer Cells | ||
|---|---|---|---|
| Genome Stability | Chemotherapy and radiation therapy induction | ||
 | Exogenous (chemical, physical, biological agents) Endogenous (DNA replication errors, chromosomal segregation defects) |  | Doxorubicin, etoposide, et al.
|
| Telomere depletion |  | ||
 | Cell replication (telomere loss) Telomere dysfunction (DNA damage) |  | |
 | |||
| Epigenetic changes | Oncogenic signaling | ||
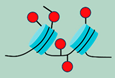 | DNA methylation patterns, Post-translational modifications Chromatin remodeling non coding RNA (ncRNA) |  | RAS, MYC, BRAF and PTEN, et al induced cancer cell senescence |
| Metabolic changes | Immune cytokine induction | ||
 | Glucose metabolism Amino acid metabolism Fatty acid metabolism ATP, NAD+ |  | Immune factors produced by immune cells induce cancer cell senescence (IFN-γ, TNF) |
| Signaling Pathway | Hypoxia and reactive oxygen species induction | ||
 | PARP1/AMPK pathway ROS/AMPK/mTOR pathway SIRTs mediated signaling pathways |  | Hypoxic conditions can induce upregulation of HIF-1 α, thereby inducing cancer cell senescence |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Gao, Y.; Zhang, S.; Wang, Y.; Du, Y.; Hao, S.; Ni, T. The Regulation of Cellular Senescence in Cancer. Biomolecules 2025, 15, 448. https://doi.org/10.3390/biom15030448
Zhang X, Gao Y, Zhang S, Wang Y, Du Y, Hao S, Ni T. The Regulation of Cellular Senescence in Cancer. Biomolecules. 2025; 15(3):448. https://doi.org/10.3390/biom15030448
Chicago/Turabian StyleZhang, Xianhong, Yue Gao, Siyu Zhang, Yixiong Wang, Yitian Du, Shuailin Hao, and Ting Ni. 2025. "The Regulation of Cellular Senescence in Cancer" Biomolecules 15, no. 3: 448. https://doi.org/10.3390/biom15030448
APA StyleZhang, X., Gao, Y., Zhang, S., Wang, Y., Du, Y., Hao, S., & Ni, T. (2025). The Regulation of Cellular Senescence in Cancer. Biomolecules, 15(3), 448. https://doi.org/10.3390/biom15030448