
Oral and Non-Oral Cholesterol-Lowering Drugs with PCSK9 and Other Biomolecules as Targets: Present Status and Future Prospects

Abstract

1. Introduction
1.1. Cholesterol Background and Function
1.2. Cholesterol Level and Carrier Lipoproteins
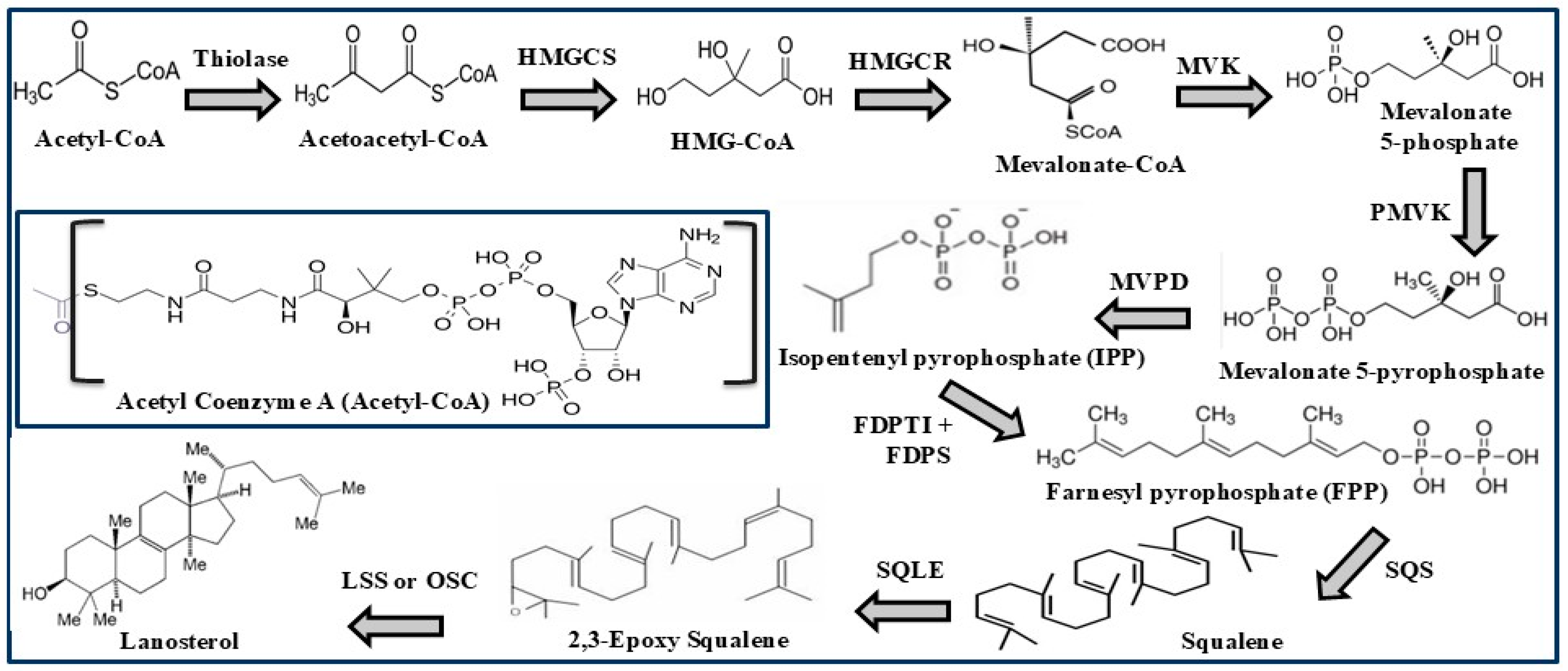
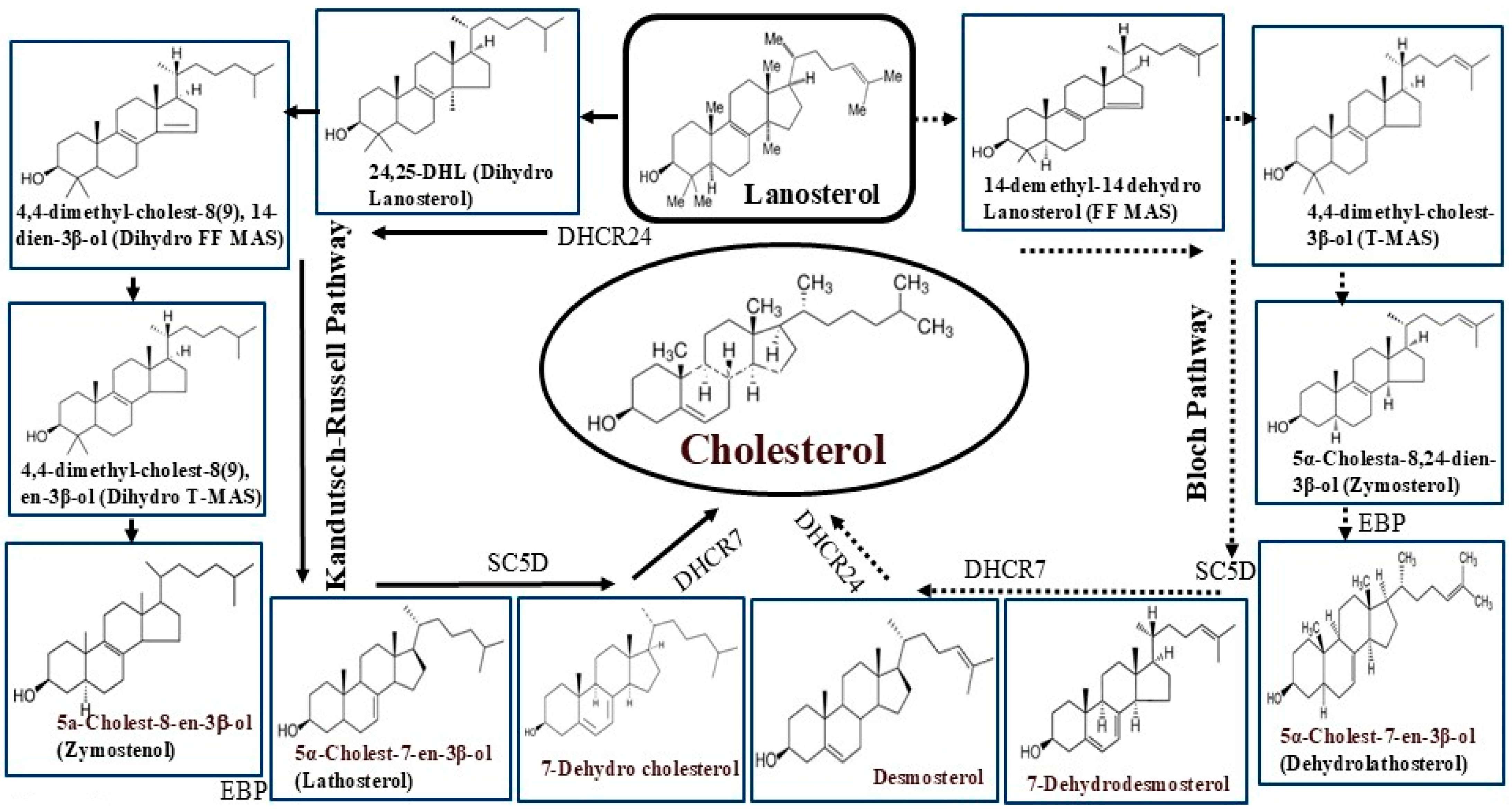
1.3. Cholesterol Biosynthesis and Dietary Sources
1.4. Cholesterol Pathology and Management
2. Biomolecules as Targets for Lowering Cholesterol
2.1. HMG CoA Reductase (HMGCR)
2.2. PCSK9
2.3. ATP-Citrate Lyase
2.4. Squalene Synthase (SQS)
2.5. Squalene Epoxidase (SQLE)
2.6. Oxidosqualene Cyclase (OSC)
2.7. Emopamil Binding Protein (EBP) or 3β-Hydroxysteroid Δ8-Δ7 Isomerase
2.8. Bile Acids
2.9. Cholesterol Absorption
2.10. Lipoproteins
3. Drugs Approved for Lowering Cholesterol
3.1. Oral Drugs
3.1.1. Statins
3.1.2. Non-statins
3.2. Nonoral (Injectable) Drugs
3.2.1. Alirocumab (Praluent)
3.2.2. Evolocumab (Repatha)
3.2.3. Inclisiran (Leqvio)
3.2.4. Mipomersen
3.2.5. Lomitapide
3.2.6. LIB003 (Lerodalcibep)
4. Oral PCSK9 Drugs in Progress
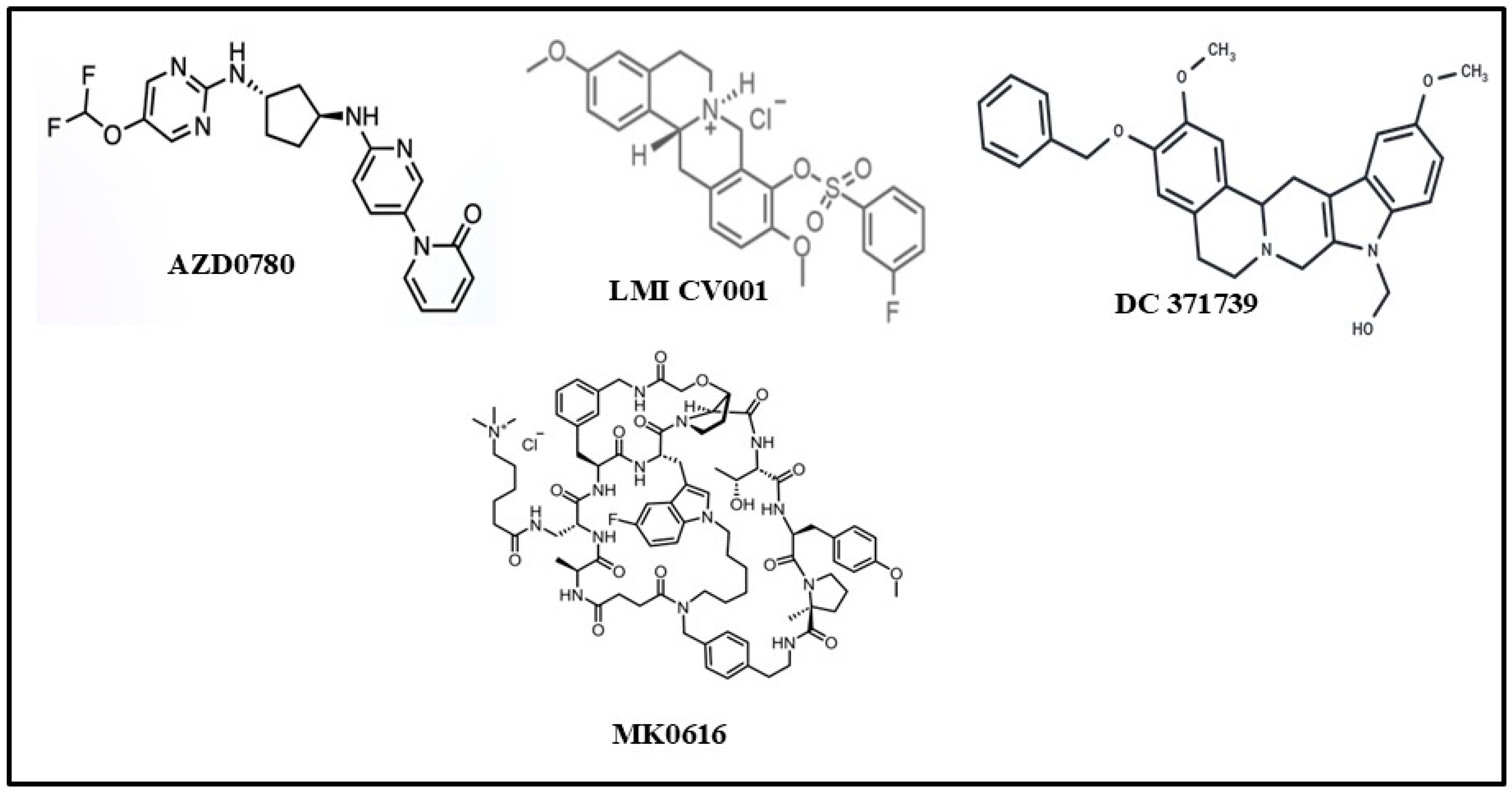
4.1. AZD0780 (Laroprovstat)
4.2. AZD8233
4.3. NNC0385-0434
4.4. CVI-LM001
4.5. DC371739
4.6. MK-0616 (Enlicitide Chloride)
4.7. PCSK9-Derived Peptides
5. Plant Sterols as Cholesterol-Reducing Agents
6. Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | Amino acid |
| ACLY | Adenosine triphosphate-citrate lyase |
| ADP | Adenosine Diphosphate |
| CVD | Cardiovascular disease |
| CHD | Congenital heart disease |
| Da | Dalton |
| DALYS | Disability-Adjusted Life Years |
| DHCR24 | Dehydrocholesterol reductase24 |
| EBP | Emopamil binding protein |
| EGF-A | Epidermal Growth Factor like-domain A |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| GI | Gastrointestinal |
| h | Human |
| HMG-CoA | 3-Hydroxy-3-Methyl Glutaryl Coenzyme A |
| IDL | Intermediate-density lipoprotein |
| IR | Immediate Release |
| LDL | Low-density lipoprotein |
| mAb | Monoclonal antibody |
| MHRA | Medicines and Healthcare products Regulatory Agency, UK |
| mRNA | Messenger Ribonucleic Acid |
| MW | Molecular weight |
| OSC | Oxidosqualene cyclase |
| PCSK9 | Proprotein Convertase Subtilisin Kexin9 |
| RISC | RNA-induced silencing complex |
| SNAC | Sodium N-(8-[2-hydroxybenzoyl] amino) caprylate |
| SQLE | Squalene epoxidase |
| SQS | Squalene synthase |
| tBu | Tertiary butyl |
| THPBs | Tetrahydroproto-berberines |
| VLDL | Very low-density lipoprotein |
References
- Kuijper, P.M.J.C. History in medicine: The story of cholesterol, lipids and cardiology. e-J. Cardiol. Pract. (Eur. Soc. Cardiol.) 2021, 19, 1–12. [Google Scholar]
- Huff, T.; Boyd, B.; Jialal, I. Physiology, Cholesterol. [Updated 6 March 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470561/ (accessed on 11 March 2025).
- Schade, D.S.; Shey, L.; Eaton, R.P. Cholesterol Review: A Metabolically Important Molecule. Endocr. Pract. 2020, 26, 1514–1523. [Google Scholar]
- Zampelas, A.; Magriplis, E. New Insights into Cholesterol Functions: A Friend or an Enemy? Nutrients 2019, 11, 1645. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Gong, K.; Zhang, F.; Meng, X.; Han, J. Regulation of cholesterol homeostasis in health and diseases: From mechanisms to targeted therapeutics. Sig. Transduct. Target Ther. 2022, 7, 1–29. [Google Scholar]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [PubMed]
- Idoko, A.; Ugwudike, P.O.; Ayomide, T.A.; Blessing, N.O. Cholesterol and its implications. Univers. J. Pharm. Research. 2020, 5, 52–63. [Google Scholar]
- Ginsberg, H.N. Lipoprotein Physiology. Endocrinol. Metab. Clin. N. Am. 1998, 27, 503–518. [Google Scholar]
- Tulenko, T.N.; Sumner, A.E. The physiology of lipoproteins. J. Nucl. Cardiol. 2002, 9, 638–649. [Google Scholar]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. [Updated 14 January 2024]. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK305896/ (accessed on 7 February 2025).
- Daniels, T.F.; Killinger, K.M.; Michal, J.J.; Wright, R.W., Jr.; Jiang, Z. Lipoproteins, cholesterol homeostasis and cardiac health. Int. J. Biol. Sci. 2009, 5, 474–488. [Google Scholar]
- Soppert, J.; Lehrke, M.; Marx, N.; Jankowski, J.; Noels, H. Lipoproteins and lipids in cardiovascular disease: From mechanistic insights to therapeutic targeting. Adv. Drug Deliv. Rev. 2020, 159, 4–33. [Google Scholar]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef]
- Cerqueira, N.M.F.S.A.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Pathway Summary for Pathway SMP0121057, Bloch Pathway (Cholesterol Biosynthesis). Source: PathBank. Available online: https://pubchem.ncbi.nlm.nih.gov/pathway/PathBank:SMP0121057 (accessed on 7 February 2025).
- Mazein, A.; Watterson, S.; Hsieh, W.-Y.; Griffiths, W.J.; Ghazal, P. A comprehensive machine-readable view of the mammalian cholesterol biosynthesis pathway. Biochem. Pharmacol. 2013, 86, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yutuc, E.; Griffiths, W.J. Cholesterol metabolism pathways are the intermediates more important than the products? FEBS J. 2021, 288, 3727–3745. [Google Scholar] [CrossRef]
- Schoeneck, M.; Iggman, D. The effects of foods on LDL cholesterol levels: A systematic review of the accumulated evidence from systematic reviews and meta-analyses of randomized controlled trials. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1325–1338. [Google Scholar] [CrossRef]
- Carson, J.A.S.; Lichtenstein, A.H.; Anderson, C.A.M.; Appel, L.J.; Kris-Etherton, P.M.; Meyer, K.A.; Petersen, K.; Polonsky, T.; Van Horn, L. Dietary Cholesterol and Cardiovascular Risk: A Science Advisory from the American Heart Association. Circulation 2020, 141, e39–e53. [Google Scholar] [CrossRef]
- Soliman, G.A. Dietary Cholesterol and the Lack of Evidence in Cardiovascular Disease. Nutrients 2018, 10, 780. [Google Scholar] [CrossRef] [PubMed]
- Wadhera, R.K.; Steen, D.L.; Khan, I.; Giugliano, R.P.; Foody, J.M. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J. Clin. Lipidol. 2016, 10, 472–489. [Google Scholar] [CrossRef]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-induced toxicity: An integrated view of the role of cholesterol in multiple diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef]
- Moutzouri, E.; Elisaf, M.; Liberopoulos, E.N. Hypocholesterolemia. Curr. Vasc. Pharmacol. 2011, 9, 200–212. [Google Scholar] [CrossRef]
- Nago, N.; Ishikawa, S.; Goto, T.; Kayaba, K. Low cholesterol is associated with mortality from stroke, heart disease, and cancer: The Jichi Medical School Cohort Study. J. Epidemiol. 2011, 21, 67–74. [Google Scholar]
- Nagar, M. Health risks of very low cholesterol. Sci. J. Lander Coll. Arts Sci. 2011, 5, 26–39. [Google Scholar]
- Available online: https://www.who.int/data/gho/indicator-metadata-registry/imr-details/3236#:~:text=Globally%2C%20a%20third%20of%20ischaemic,or%202%25%20of%20total%20DALYS (accessed on 7 February 2025).
- Di Cesare, M.; Perel, P.; Taylor, S.; Kabudula, C.; Bixby, H.; Gaziano, T.A.; McGhie, D.V.; Mwangi, J.; Pervan, B.; Narula, J.; et al. The Heart of the World. Glob. Heart 2024, 19, 11. [Google Scholar] [CrossRef]
- Lindstrom, M.; DeCleene, N.; Dorsey, H.; Fuster, V.; Johnson, C.O.; LeGrand, K.E.; Mensah, G.A.; Razo, C.; Stark, B.; Turco, J.V.; et al. Global Burden of Cardiovascular Diseases and Risks Collaboration, 1990–2021. J. Am. Coll. Cardiol. 2022, 80, 2372–2425. [Google Scholar] [PubMed]
- Jiang, S.Y.; Li, H.; Tang, J.J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.-K.; Shi, X.-J.; Cui, H.-W.; Tang, J.; et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat. Commun. 2018, 9, 5138. [Google Scholar]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5, 248. [Google Scholar]
- Tobert, J. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [PubMed]
- Bansal, A.B.; Cassagnol, M. HMG-CoA Reductase Inhibitors. [Updated 3 July 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK542212/ (accessed on 11 March 2025).
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar]
- Adhyaru, B.B.; Jacobson, T.A. Safety and efficacy of statin therapy. Nat. Rev. Cardiol. 2018, 15, 757–769. [Google Scholar]
- de Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar]
- Chou, R.; Cantor, A.; Dana, T.; Wagner, J.; Ahmed, A.Y.; Fu, R.; Ferencik, M. Statin Use for the Primary Prevention of Cardiovascular Disease in Adults Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2022, 328, 754–771. [Google Scholar]
- Fitchett, D.H.; Hegele, R.A.; Verma, S. Statin Intolerance. Circulation 2015, 131, e389–e391. [Google Scholar] [PubMed]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Pratt, A.; Chrétien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [PubMed]
- Basak, A.; Palmer-Smith, H.; Mishra, P. Proprotein Convertase Subtilisin Kexin9 (PCSK9): A Novel Target for Cholesterol Regulation. Prot. Pep. Lett. 2012, 19, 575–585. [Google Scholar]
- Bao, X.; Liang, Y.; Chang, H.; Cai, T.; Feng, B.; Gordon, K.; Zhu, Y.; Shi, H.; He, Y.; Xie, L. Targeting proprotein convertase subtilisin/kexin type 9 (PCSK9): From bench to bedside. Sig. Transduct. Target Ther. 2024, 9, 1–49. [Google Scholar]
- Lagace, T.A. PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar]
- Libby, P.; Tokgözoğlu, L. Chasing LDL cholesterol to the bottom-PCSK9 in perspective. Nat. Cardiovasc. Res. 2022, 1, 554–561. [Google Scholar]
- Seidah, N.G.; Prat, A. The Multifaceted Biology of PCSK9. Endocr. Rev. 2022, 43, 558–582. [Google Scholar] [CrossRef]
- Lemus, H.N.; Mendivil, C.O. Adenosine triphosphate citrate lyase: Emerging target in the treatment of dyslipidemia. J. Clin. Lipidol. 2015, 9, 384–389. [Google Scholar]
- Groot, P.H.E.; Pearce, N.J.; Gribble, A.D. ATP-citrate Lyase: A Potential Target for Hypolipidemic Intervention. Curr. Med. Chem.-Immunol. Endocr. Metab. Agents 2003, 3, 211–217. [Google Scholar] [CrossRef]
- Dolle, R.E.; McNair, D.; Hughes, M.J.; Kruse, L.I.; Eggelston, D.; Saxty, B.A.; Wells, T.N.; Groot, P.H. ATP-citrate lyase as a target for hypolipidemic intervention. Sulfoximine and 3-hydroxy-beta-lactam containing analogues of citric acid as potential tight-binding inhibitors. J. Med. Chem. 1992, 35, 4875–4884. [Google Scholar]
- Charlton-Menys, V.; Durrington, P.N. Squalene synthase inhibitors: Clinical pharmacology and cholesterol-lowering potential. Drugs 2007, 67, 11–16. [Google Scholar]
- Chua, N.K.; Coates, H.W.; Brown, A.J. Squalene monooxygenase: A journey to the heart of cholesterol synthesis. Prog. Lipid Res. 2020, 79, 101033. [Google Scholar]
- Huff, M.W.; Telford, D.E. Lord of the rings—The mechanism for oxidosqualene:lanosterol cyclase becomes crystal clear. Trends Pharmacol. Sci. 2005, 26, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Debler, E.W.; Li, X. Structural enzymology of cholesterol biosynthesis and storage. Curr. Opin. Struct. Biol. 2022, 74, 102369. [Google Scholar]
- Sato, R. Recent advances in regulating cholesterol and bile acid metabolism. Biosci. Biotechnol. Biochem. 2020, 84, 2185–2192. [Google Scholar] [PubMed]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009, 32 (Suppl. 2), S237–S245. [Google Scholar]
- Fleishman, J.S.; Kumar, S. Bile acid metabolism and signaling in health and disease: Molecular mechanisms and therapeutic targets. Signal Transduct. Target Ther. 2024, 9, 1–51. [Google Scholar]
- Hofmann, A.F. Bile Acids: The Good, the Bad, and the Ugly. News Physiol. Sci. 1999, 14, 24–29. [Google Scholar] [CrossRef]
- Ostlund, R.E., Jr. Cholesterol absorption. Curr. Opin. Gastroenterol. 2002, 18, 254–258. [Google Scholar]
- Andrade, I.; Santos, L.; Ramos, F. Chapter 5—An Overview of Cholesterol Absorption. In The Molecular Nutrition of Fats; Patel, V.B., Ed.; Elsevier: New York, NY, USA, 2019; pp. 65–76. [Google Scholar]
- Leitersdorf, E. Cholesterol absorption inhibition: Filling an unmet need in lipid-lowering management. Eur. Heart J. Suppl. 2001, 3, E17–E23. [Google Scholar] [CrossRef]
- Goldberg, A.C. Novel therapies and new targets of treatment for familial hypercholesterolemia. J. Clin. Lipidol. 2010, 4, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Kounatidis, D.; Vallianou, N.G.; Poulaki, A.; Evangelopoulos, A.; Panagopoulos, F.; Stratigou, T.; Geladari, E.; Karampela, I.; Dalamaga, M. ApoB100 and Atherosclerosis: What’s New in the 21st Century? Metabolites 2024, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Nijstad, N.; Franceschini, L. Regulation of microsomal triglyceride transfer protein. Clin Lipidol. 2011, 6, 293–303. [Google Scholar] [CrossRef]
- Last, A.R.; Ference, J.D.; Menzel, E.R. Hyperlipidemia: Drugs for Cardiovascular Risk Reduction in Adults. Am Fam Physician. 2017, 95, 78–87. [Google Scholar]
- McPherson, R.; Adreak, N.; Sharma, A. Medications for Lipid Control: Statins vs Newer Drugs. Can. J. Cardiol. 2024, 40, S26–S34. [Google Scholar] [CrossRef]
- Ma, W.; Pan, Q.; Pan, D.; Xu, T.; Zhu, H.; Li, D. Efficacy and Safety of Lipid-Lowering Drugs of Different Intensity on Clinical Outcomes: A Systematic Review and Network Meta-Analysis. Front. Pharmacol. 2021, 12, 713007. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Catapano, A.L. The pharmacology of cholesterol-lowering drugs. Eur. Atheroscler. J. 2022, 1, 2–13. [Google Scholar]
- Sizar, O.; Khare, S.; Patel, P.; Talati, R. Statin Medications. [Updated 29 February 2024]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430940/ (accessed on 11 March 2025).
- Pignone, M.; Cannon, C.P. Low-density lipoprotein cholesterol-lowering therapy in the primary prevention of cardiovascular disease. UpToDate 2023, 16, 1–26. [Google Scholar]
- Mangione, C.M. Statin Use for the Primary Prevention of Cardiovascular Disease in Adults. JAMA. 2022, 328, 746–753. [Google Scholar]
- Raal, F.J.; Mohamed, F. Statins: Are they appropriate for all patients? Lancet Glob. Health 2022, 10, e305–e306. [Google Scholar] [PubMed]
- Ramkumar, S.; Raghunath, A.; Raghunath, S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol. Sin. 2016, 32, 631–639. [Google Scholar]
- Statins Market Size to Hit US $22 Billion by 2032, Owing to Increasing Investments in Healthcare Sector Globally. Persistence Market Research. 2 March 2023. Available online: https://www.globenewswire.com/news-release/2023/03/02/2619166/0/en/Statins-Market-Size-To-Hit-US-22-Billion-By-2032-Owing-To-Increasing-Investments-in-Healthcare-Sector-Globally-Persistence-Market-Research.html (accessed on 7 February 2025).
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar]
- Climent, E.; Benaiges, D.; Pedro-Botet, J. Hydrophilic or Lipophilic Statins? Front. Cardiovasc. Med. 2021, 8, 687585. [Google Scholar]
- Lin, S.-Y.; Baumann, K.; Zhou, C.; Zhou, W.; Cuellar, A.E.; Xue, H. Trends in Use and Expenditures for Brand-name Statins After Introduction of Generic Statins in the US, 2002–2018. JAMA Netw. Open 2021, 4, e2135371. [Google Scholar]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clin. (Barc) 2018, 150, 398–402. [Google Scholar]
- Kulkarni, S.; Watts, M.M.; Kostapanos, M. Statins. BMJ 2024, 384, 1–10. [Google Scholar]
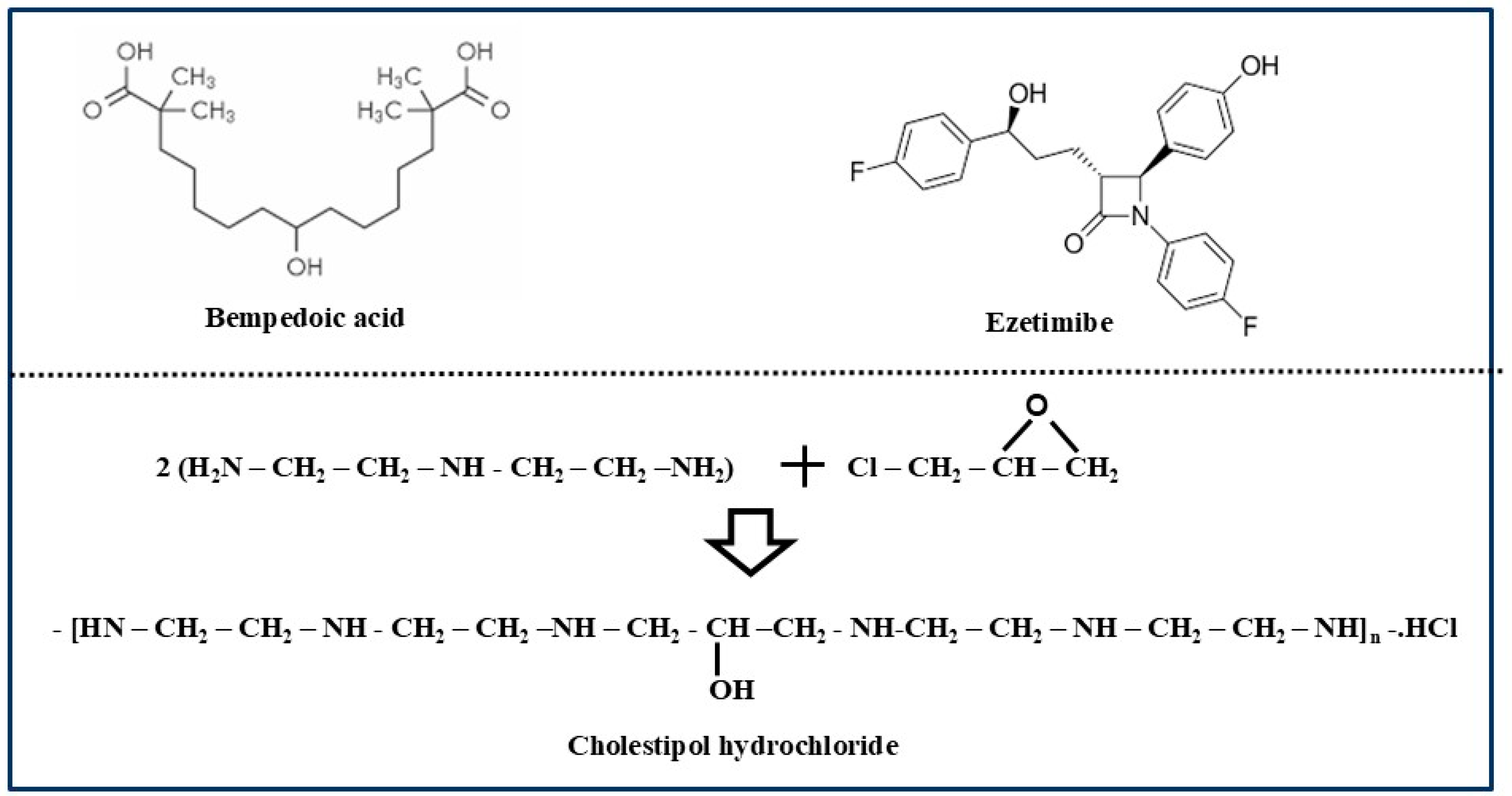
- Markham, A. Bempedoic Acid: First Approval. Drugs 2020, 80, 747–753. [Google Scholar] [PubMed]
- Kulshreshtha, M. An Update on New Cholesterol Inhibitor: Bempedoic Acid. Curr. Cardiol. Rev. 2022, 18, e141221198875. [Google Scholar]
- Nissen, S.E.; Lincoff, M.A.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.P.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. for CLEAR Outcomes Investigators. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N. Engl. J. Med. 2023, 380, 1353–1364. [Google Scholar]
- Sizar, O.; Nassereddin, A.; Talati, R. Ezetimibe. [Updated 28 August 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532879/ (accessed on 11 March 2025).
- Clader, J.W. The discovery of ezetimibe: A view from outside the receptor. J. Med. Chem. 2004, 47, 1–9. [Google Scholar] [PubMed]
- Phan, B.A.P.; Dayspring, T.D.; Toth, P.P. Ezetimibe therapy: Mechanism of action and clinical update. Vasc. Health Risk Manag. 2012, 8, 415–427. [Google Scholar] [PubMed]
- Staels, B.; Handelsman, Y.; Fonseca, V. Bile Acid Sequestrants for Lipid and Glucose Control. Curr. Diab. Rep. 2010, 10, 70–77. [Google Scholar]
- Colestipol. [Updated 28 September 2017]. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548432/ (accessed on 11 March 2025).
- Heel, R.C.; Brogden, R.N.; Pakes, G.E.; Speight, T.M.; Avery, G.S. Colestipol: A review of its pharmacological properties and therapeutic efficacy in patients with hypercholesterolaemia. Drugs 1980, 19, 161–180. [Google Scholar]
- Patel, P.H.; Can, A.S. Colesevelam. [Updated 1 May 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557815/ (accessed on 11 March 2025).
- Bays, H.; Jones, P.H. Colesevelam hydrochloride: Reducing atherosclerotic coronary heart disease risk factors. Vasc. Health Risk Manag. 2007, 3, 733–742. [Google Scholar] [PubMed]
- Radosh, L. Colesevelam (WelChol) for Reduction of LDL Cholesterol. Am. Fam. Phys. 2005, 72, 321–324. [Google Scholar]
- Riaz, S.; John, S. Cholestyramine Resin. [Updated 8 May 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK534089/ (accessed on 11 March 2025).
- Scaldaferri, F.; Pizzoferrato, M.; Ponziani, F.R.; Gasbarrini, G.; Gasbarrini, A. Use and indications of cholestyramine and bile acid sequestrants. Intern. Emerg. Med. 2013, 8, 205–210. [Google Scholar]
- Nishimoto, T.; Amano, Y.; Tozawa, R.; Ishikawa, E.; Imura, Y.; Yukimasa, H.; Sugiyama, Y. Lipid-lowering properties of TAK-475, a squalene synthase inhibitor, in vivo and in vitro. Br. J. Pharmacol. 2003, 139, 911–918. [Google Scholar]
- Liu, C.-I.; Jeng, W.-Y.; Chang, W.-J.; Ko, T.-P.; Wang, A.H.-J. Binding Modes of Zaragozic Acid A to Human Squalene Synthase and Staphylococcal Dehydrosqualene Synthase. J. Biol. Chem. 2012, 287, 18750–18757. [Google Scholar]
- Chugh, A.; Ray, A.; Gupta, J.B. Squalene epoxidase as hypocholesterolemic drug target revisited. Prog. Lipid Res. 2003, 42, 37–50. [Google Scholar]
- Belter, A.; Skupinska, M.; Giel-Pietraszuk, M.; Grabarkiewicz, T.; Rychlewski, L.; Barciszewski, J. Squalene monooxygenase—A target for hypercholesterolemic therapy. Biol. Chem. 2011, 392, 1053–1075. [Google Scholar] [CrossRef] [PubMed]
- Hajar, R. PCSK 9 Inhibitors: A Short History and a New Era of Lipid-lowering Therapy. Heart Views 2019, 20, 74–75. [Google Scholar] [CrossRef]
- Coppinger, C.; Movahed, M.R.; Azemawah, V.; Peyton, L.; Gregory, J.; Hashemzadeh, M.A. Comprehensive Review of PCSK9 Inhibitors. J. Cardiovasc. Pharmacol. Ther. 2022, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Agnello, F.; Mauro, M.S.; Rochira, C.; Landolina, D.; Finocchiaro, S.; Greco, A.; Ammirabile, N.; Raffo, C.; Mazzone, P.M.; Spagnolo, M.; et al. PCSK9 inhibitors: Current status and emerging frontiers. Expert Rev. Cardiovasc. Ther. 2023, 23, 1–18. [Google Scholar]
- Raedler, L.A. Praluent (Alirocumab): First PCSK9 Inhibitor Approved by the FDA for Hypercholesterolemia. Am. Health Drug Benefits 2016, 9, 123–126. [Google Scholar]
- Manniello, M.; Pisano, M. Alirocumab (Praluent): First in the New Class of PCSK9 Inhibitors. Pharm. Ther. 2016, 41, 28–53. [Google Scholar]
- Fala, L. Repatha (Evolocumab): Second PCSK9 Inhibitor Approved by the FDA for Patients with Familial Hypercholesterolemia. Am. Health Drug Benefits 2016, 9, 136–139. [Google Scholar]
- McDonagh, M.; Peterson, K.; Holzhammer, B.; Fazio, S. A Systematic Review of PCSK9 Inhibitors Alirocumab and Evolocumab. J. Manag. Care Spec. Pharm. 2016, 22, 641–653. [Google Scholar] [PubMed]
- Stein, E.A.; Mellis, S.; Yancopoulos, G.D.; Stahl, N.; Logan, D.; Smith, W.B.; Lisbon, E.; Gutierrez, M.; Webb, C.; Wu, R.; et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 2012, 366, 1108–1118. [Google Scholar] [CrossRef]
- Dias, C.S.; Shaywitz, A.J.; Wasserman, S.M.; Smith, B.P.; Gao, B.; Stolman, D.S.; Crispino, C.P.; Smirnakis, K.V.; Emery, M.G.; Colbert, A.; et al. Effects of AMG145 on low-density lipoprotein cholesterol levels: Results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J. Am. Coll. Cardiol. 2012, 60, 1888–1898. [Google Scholar] [CrossRef]
- Ridker, P.M.; Tardif, J.C.; Amarenco, P.; Duggan, W.; Glynn, R.J.; Jukema, J.W.; Kastelein, J.J.P.; Kim, A.M.; Koenig, W.; Nissen, S.; et al. Lipid-Reduction Variability and Antidrug-Antibody Formation with Bococizumab. N. Engl. J. Med. 2017, 376, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Adams, B. Pfizer Dumps PCSK9 Inhibitor Bococizumab After Finding ’no Value’ in Drug. Fierce Biotech Questex. 1 November 2016. Available online: https://www.fiercebiotech.com/biotech/pfizer-dumps-pcsk9i-inhibitor-bococizumab-after-finding-no-value-med (accessed on 7 February 2025).
- Ray, K.K.; Troquay, R.P.T.; Visseren, F.L.J.; Leiter, L.A.; Wright, R.S.; Vikarunnessa, S.; Talloczy, Z.; Zang, X.; Maheux, P.; Lesogor, A.; et al. Long-term efficacy and safety of inclisiran in patients with high cardiovascular risk and elevated LDL cholesterol (ORION-3): Results from the 4-year open-label extension of the ORION-1 trial. Lancet Diabetes Endocrinol. 2023, 11, 109–119. [Google Scholar] [PubMed]
- Merćep, I.; Friščić, N.; Strikić, D.; Reiner, Ž. Advantages and Disadvantages of Inclisiran: A Small Interfering Ribonucleic Acid Molecule Targeting PCSK9-A Narrative Review. Cardiovasc. Ther. 2022, 2022, 8129513. [Google Scholar] [PubMed]
- Wilkinson, M.J.; Bajaj, A.; Brousseau, M.E.; Taub, P.R. Harnessing RNA Interference for Cholesterol Lowering: The Bench-to-Bedside Story of Inclisiran. J. Am. Heart Assoc. 2024, 13, e032031. [Google Scholar] [CrossRef]
- Desai, N.R.; Campbell, C.; Electricwala, B.; Petrou, M.; Trueman, D.; Woodcock, F.; Cristino, J. Cost Effectiveness of Inclisiran in Atherosclerotic Cardiovascular Patients with Elevated Low-Density Lipoprotein Cholesterol Despite Statin Use: A Threshold Analysis. Am. J. Cardiovasc. Drugs 2022, 22, 545–556. [Google Scholar]
- Nandakumar, R.; Matveyenko, A.; Thomas, T.; Pavlyha, M.; Ngai, C.; Holleran, S.; Ramakrishnan, R.; Ginsberg, H.N.; Karmally, W.; Marcovina, S.M.; et al. Effects of mipomersen, an apolipoprotein B100 antisense, on lipoprotein (a) metabolism in healthy subjects. J. Lipid Res. 2018, 59, 2397–2402. [Google Scholar]
- Visser, M.E.; Wagener, G.; Baker, B.F.; Geary, R.S.; Donovan, J.M.; Beuers, U.H.; Nederveen, A.J.; Verheij, J.; Trip, M.D.; Basart, D.C.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: A randomized, double-blind, placebo-controlled trial. Eur. Heart J. 2012, 33, 1142–1149. [Google Scholar]
- Hamada, M.; Farzam, K. Mipomersen. [Updated 2 July 2023]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK592392/ (accessed on 11 March 2025).
- Suppressa, P.; Coppola, C.; Cocco, V.; O’Brien, S. Long-term effectiveness and safety of lomitapide in patients with homozygous familial hypercholesterolemia: An observational case series. Orphanet J. Rare Dis. 2024, 19, 370. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, L.; Wang, X.; Jin, H. RNA-based therapeutics: An overview and prospectus. Cell Death Dis. 2022, 13, 644. [Google Scholar]
- Raal, F.; Fourie, N.; Scott, R.; Blom, D.; De Vries Basson, M.; Kayikcioglu, M.; Caldwell, K.; Kallend, D.; Stein, E. LIBerate-HeFH investigators. Long-term efficacy and safety of lerodalcibep in heterozygous familial hypercholesterolaemia: The LIBerate-HeFH trial. Eur. Heart J. 2023, 44, 4272–4280. [Google Scholar]
- Raal, F.; Mehta, V.; Kayikcioglu, M.; Blom, D.; Gupta, P.; Daniels, C.; Vest, J.; Caldwell, K.; Bahassi, E.M.; Kallend, D.; et al. Randomized, open-label, cross-over, phase-3 study to evaluate efficacy and safety of LIB003 compared with evolocumab in homozygous familial hypercholesterolaemia patients on stable lipid-lowering therapy (liberate-HOFH). Atherosclerosis 2023, 379, S24–S25. [Google Scholar]
- Thompson, G. Limitations of cholesterol lowering with PCSK9 inhibitors. Lancet Diabetes Endocrinol. 2017, 5, 241–243. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Skavdis, A.; Sourlas, A.; Papakonstantinou, E.J.; Peña Genao, E.; Echavarria Uceta, R.; Guzman, E. Safety and Tolerability of PCSK9 Inhibitors: Current Insights. Clin. Pharmacol. 2020, 12, 191–202. [Google Scholar] [PubMed]
- Zhen, F.; Li, X.; Kei, W.; Tong, W.K.; He, Q.; Xiao, Z.; Xiang, X.; Tang, Z. Real-world safety of PCSK9 inhibitors: A pharmacovigilance study based on spontaneous reports in FAERS. Front. Pharmacol. 2022, 13, 1–11. [Google Scholar]
- Ferri, N.; Marodin, G. Emerging oral therapeutic strategies for inhibiting PCSK9. Atheroscler. Plus 2024, 59, 25–31. [Google Scholar]
- Vega, R.B.; Garkaviy, P.; Knöchel, J.; Barbour, A.; Rudvik, A.; Laru, J.; Twaddle, L.; Mccarthy, M.C.; Birgitte Rosenmeier, J. AZD0780, the first oral small molecule PCSK9 inhibitor for the treatment of hypercholesterolemia: Results from a randomized, single-blind, placebo-controlled phase 1 trial. Atherosclerosis 2024, 395, 118514. [Google Scholar]
- Burnett, J.R.; Hooper, A.J. MK-0616: An oral PCSK9 inhibitor for hypercholesterolemia treatment. Expert Opin. Investig. Drugs 2023, 32, 873–878. [Google Scholar]
- Available online: https://www.pharmaceutical-technology.com/data-insights/azd-0780-astrazeneca-cardiovascular-disease-likelihood-of-approval/ (accessed on 7 February 2025).
- A Study to Assess the Efficacy, Safety and Tolerability of Different Doses of AZD0780 in Patients with Dyslipidemia (PURSUIT). Available online: https://clinicaltrials.gov/study/NCT06173570?rank=1 (accessed on 7 February 2025).
- Gennemark, P.; Walter, K.; Clemmensen, N.; Rekic, D.; Nilsson, C.A.M.; Knochel, J.; Hölttä, M.; Wernevik, L.; Rosengren, B.; Kakol-Palm, D.; et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med. 2021, 13, eabe9117. [Google Scholar]
- Li, X.Q.; Elebring, M.; Dahlén, A.; Weidolf, L. In Vivo Metabolite Profiles of an N-Acetylgalactosamine-Conjugated Antisense Oligonucleotide AZD8233 Using Liquid Chromatography High-Resolution Mass Spectrometry: A Cross-Species Comparison in Animals and Humans. Drug Metab. Dispos. 2023, 51, 1350–1361. [Google Scholar] [CrossRef]
- Mohamed, F.; Mansfield, B.; Raal, F.J. Targeting PCSK9 and Beyond for the Management of Low-Density Lipoprotein Cholesterol. J. Clin. Med. 2023, 12, 5082. [Google Scholar] [CrossRef]
- Koren, M.J.; Descamps, O.; Hata, Y.; Hengeveld, E.M.; Hovingh, G.K.; Ikonomidis, I.; Jensen, R.J.; Langbakke, I.H.; Martens, F.M.A.C.; Søndergaard, A.L.; et al. PCSK9 inhibition with orally administered NNC0385-0434 in hypercholesterolaemia: A randomised, double-blind, placebo-controlled and active-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2024, 12, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://sciencehub.novonordisk.com/content/dam/hcpexperience/kol/en/publications/pmid_38310920/documents/koren-et-al-oral-pcsk9i-phase-2.pdf (accessed on 7 February 2025).
- Sawyer, T.K.; Biswas, K. Chapter 1, Peptide Drug Discovery Raison d’Etre: Engineering Mindset, Design Rules and Screening Tools. In Approaching the Next Inflection in Peptide Therapeutics: Attaining Cell Permeability and Oral Bioavailability; American Chemical Society: Washington, DC, USA, 2022; pp. 1–25. [Google Scholar]
- Liu, J.; Jiang, B.; Zhao, S.; Cai, S.; Xiang, D.; Huang, M.; Fang, P.; Ran, Z.; Chen, M.; Shou, Q.; et al. CVI-LM001, a first-in-class novel oral PCSK9 modulator, lowers plasma Ldl-c and reduces circulating PCSK9 in preclinical animal models and in hyperlipidemic human subjects. Circulation 2020, 142, A12579. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, J.; Yan, C.; Xi, C.; Wu, C.; Zhao, J.; Li, F.; Ding, Y.; Zhang, R.; Qi, S.; et al. Identification and evaluation of a lipid-lowering small compound in preclinical models and in a Phase I trial. Cell. Metab. 2022, 34, 667–680. [Google Scholar] [CrossRef]
- Jaitrong, M.; Boonsri, P.; Samosorn, S. Molecular Docking Studies of Berberine Derivatives as Novel Multitarget PCSK9 and HMGCR Inhibitors. SWU Sci. J. 2021, 37, 125–142. [Google Scholar]
- Ballantyne, C.M.; Banka, P.; Mendez, G.; Garcia, R. Phase 2b randomized trial of the oral PCSK9 inhibitor MK-0616. J. Am. Coll. Cardiol. 2023, 81, 1553–1564. [Google Scholar] [CrossRef]
- Watts, G.F. An anti-PCSK9 pill a day to keep cholesterol away: Next steps? Lancet Diabetes Endocrinol. 2024, 12, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.J.; Campeau, L.-C.; Banka, P.; Bautmans, A.; Bueters, T. Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low-Density Lipoprotein Receptor. Circulation 2023, 148, 144–158. [Google Scholar] [CrossRef]
- Ahmed, S.; Bhatt, S.A. Correction to Recent Update on the Development of PCSK9 Inhibitors for Hypercholesterolemia treatment. J. Med. Chem. 2023, 66, 9227–9228. [Google Scholar] [CrossRef]
- Kobayashi, N.; Nakamura, J. A novel method for the synthesis of macrocyclic peptides using arylsulfonyloxyacetic acids as N-terminus functional groups. Tetrahedron Lett. 2024, 141, 155073. [Google Scholar] [CrossRef]
- Kingwell, K. Macrocycle drugs serve up new opportunities. Nat. Rev. Drug Discov. 2023, 22, 771–773. [Google Scholar] [CrossRef]
- Alghamdi, R.H.; O’Reilly, P.; Lu, C.; Gomes, J.; Lagace, T.A.; Basak, A. LDL-R promoting activity of peptides derived from human PCSK9 catalytic domain (aa153-421): Design, synthesis and biochemical evaluation. Eur. J. Med. Chem. 2015, 92, 890–907. [Google Scholar] [PubMed]
- Alghamdi, R.H. Development of Inhibitors of Human PCSK9 as Potential Regulators of LDL-Receptor and Cholesterol. Master’s Thesis, Principal Supervisor: Ajoy Basak + co-supervisor: Tom Lagace, Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada, 2013. Available online: https://ruor.uottawa.ca/items/25aa5b31-0216-4f72-a067-41790829ea7d (accessed on 11 March 2025).
- Tombling, B.J.; Zhang, Y.; Huang, Y.-H.; Craik, D.J.; Wang, C.K. The emerging landscape of peptide-based inhibitors of PCSK9. Atherosclerosis 2021, 330, 52–60. [Google Scholar] [CrossRef]
- Agarwala, A.; Asim, R.; Ballantyne, C.M. Oral PCSK9 Inhibitors. Curr. Atheroscler. Rep. 2024, 26, 147–152. [Google Scholar] [PubMed]
- Basak, A. Peptides Derived from Human PCSK9 Catalytic Domain & Its Uses Thereof for Promoting LDL-R Activity. WO Patent WO2016/119067 A1, 4 August 2016. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016119067&redirectedID=true (accessed on 7 February 2025).
- Basak, A. Peptides Derived from Human PCSK9 Catalytic Domain & Their Uses Thereof for Promoting LDL-R Activity. US Patent US 2018/0023071 A1, 25 January 2018. [Google Scholar]
- Aqur Biosciences, Inc., Westlake Village, CA, USA. Available online: www.aqurbiosciences.com (accessed on 7 February 2025).
- Becker, M.; Staab, D.; Von Bergmann, K. Treatment of severe familial hypercholesterolemia in childhood with sitosterol and sitostanol. J. Pediatr. 1993, 122, 292–2926. [Google Scholar] [CrossRef]
- Bardolia, C.; Shah, A.N.; Turgeon, J. Emerging Non-statin Treatment Options for Lowering Low-Density Lipoprotein Cholesterol. Front. Cardiovasc. Med. 2021, 8, 789931. [Google Scholar]
- Kingwell, K. Pushing the envelope with PCSK9. Nat. Rev. Drug Discov. 2021, 20, 506. [Google Scholar]
- Katzmann, J.L.; Laufs, U. PCSK9-directed therapies: An update. Current Opinion in Lipidology. 2024, 35, 117–125. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Drug Name | Family Type | Target | Status | Delivery Route |
|---|---|---|---|---|---|
| 1 | Lovastatin IR (Mevacor) (Immediate Release) | Statins | HMG-CoAR | Approved | Oral |
| 2 | Atorvastatin (Lipitor/Caduet) | Statins | HMG-CoAR | Approved | Oral |
| 3 | Pravastatin (Pravachol) | Statins | HMG-CoAR | Approved | Oral |
| 4 | Rosuvastatin (Crestor/Ezallor) | Statins | HMG-CoAR | Approved | Oral |
| 5 | Fluvastatin (Lescol/Lescol X) | Statins | HMG-CoAR | Approved | Oral |
| 6 | Pitavastatin (Livalo/Zypitamag/Sprinkle) | Statins | HMG-CoAR | Approved | Oral |
| 7 | Simvastatin (Zocor/FloLipid/Vytorin) | Statins | HMG-CoAR | Approved | Oral |
| 8 | Lovastatin ER (Extended Release) (Altoprev) | Statins | HMG-CoAR | Approved | Oral |
| 9 | Bempedoic acid (Nexletol) | ATP-CLi | ATP-Citrate Lyase | Approved | Oral |
| 10 | Ezetimibe (Zeha) | Absorption inhibitor | Cholesterol absorption | Approved | Oral |
| 11 | Colestid (Colestipol) | Bile acid sequestrants | GI-elements | Approved | Oral |
| 12 | Welcol (Colesevelam) | Bile acid sequestrants | GI-elements | Approved | Oral |
| 13 | Prevalite (Cholestyramine) | Bile acid sequestrants | GI-elements | Approved | Oral |
| 14 | Alirocumab (Praluent) | PCSK9i | PCSK9 | Approved | Injection |
| 15 | Evolocumab (Repatha) | PCSK9i | PCSK9 | Approved | Injection |
| 16 | Inclisiran (Leqvio). | PCSK9i | PCSK9 | Approved | Injection |
| 17 | LIB003 (Lerodalcibep) | PCSK9i | PCSK9 | Close to approval | Injection |
| 18 | AZD0780 (Laroprovstat) | PCSK9i | PCSK9 | Under study | Oral |
| 19 | AZD8233 | PCSK9i | PCSK9 | Discontinued | Oral |
| 20 | MK-0616 (Enlicitide chloride) | PCSK9i | PCSK9 | Close to approval | Oral |
| 21 | NNC0385-0434 | PCSK9i | PCSK9 | Under study | Oral |
| 22 | CVI-LM001 | PCSK9i | PCSK9 | Under study | Oral |
| 23 | DC371739 | PCSK9i | PCSK9 | Under study | Oral |
| 24 | Lapaquistat acetate (TAK-475) | SQSi | SQS | Discontinued | Oral |
| 25 | Zaragozic acid | SQSi | SQS | Under study | Oral |
| 24 | Terbinafine (NB-598) | SQLEi | SQLE | Under study | Oral |
| 25 | Mipomersen | ApoBi | ApoB | Approved | Injection |
| 26 | Lomitapide | ApoBi/MTTPi | ApoB/MTTP | Approved | Injection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basak, A. Oral and Non-Oral Cholesterol-Lowering Drugs with PCSK9 and Other Biomolecules as Targets: Present Status and Future Prospects. Biomolecules 2025, 15, 468. https://doi.org/10.3390/biom15040468
Basak A. Oral and Non-Oral Cholesterol-Lowering Drugs with PCSK9 and Other Biomolecules as Targets: Present Status and Future Prospects. Biomolecules. 2025; 15(4):468. https://doi.org/10.3390/biom15040468
Chicago/Turabian StyleBasak, Ajoy. 2025. "Oral and Non-Oral Cholesterol-Lowering Drugs with PCSK9 and Other Biomolecules as Targets: Present Status and Future Prospects" Biomolecules 15, no. 4: 468. https://doi.org/10.3390/biom15040468
APA StyleBasak, A. (2025). Oral and Non-Oral Cholesterol-Lowering Drugs with PCSK9 and Other Biomolecules as Targets: Present Status and Future Prospects. Biomolecules, 15(4), 468. https://doi.org/10.3390/biom15040468