
Neurotropic Viruses as Acute and Insidious Drivers of Aging


Abstract
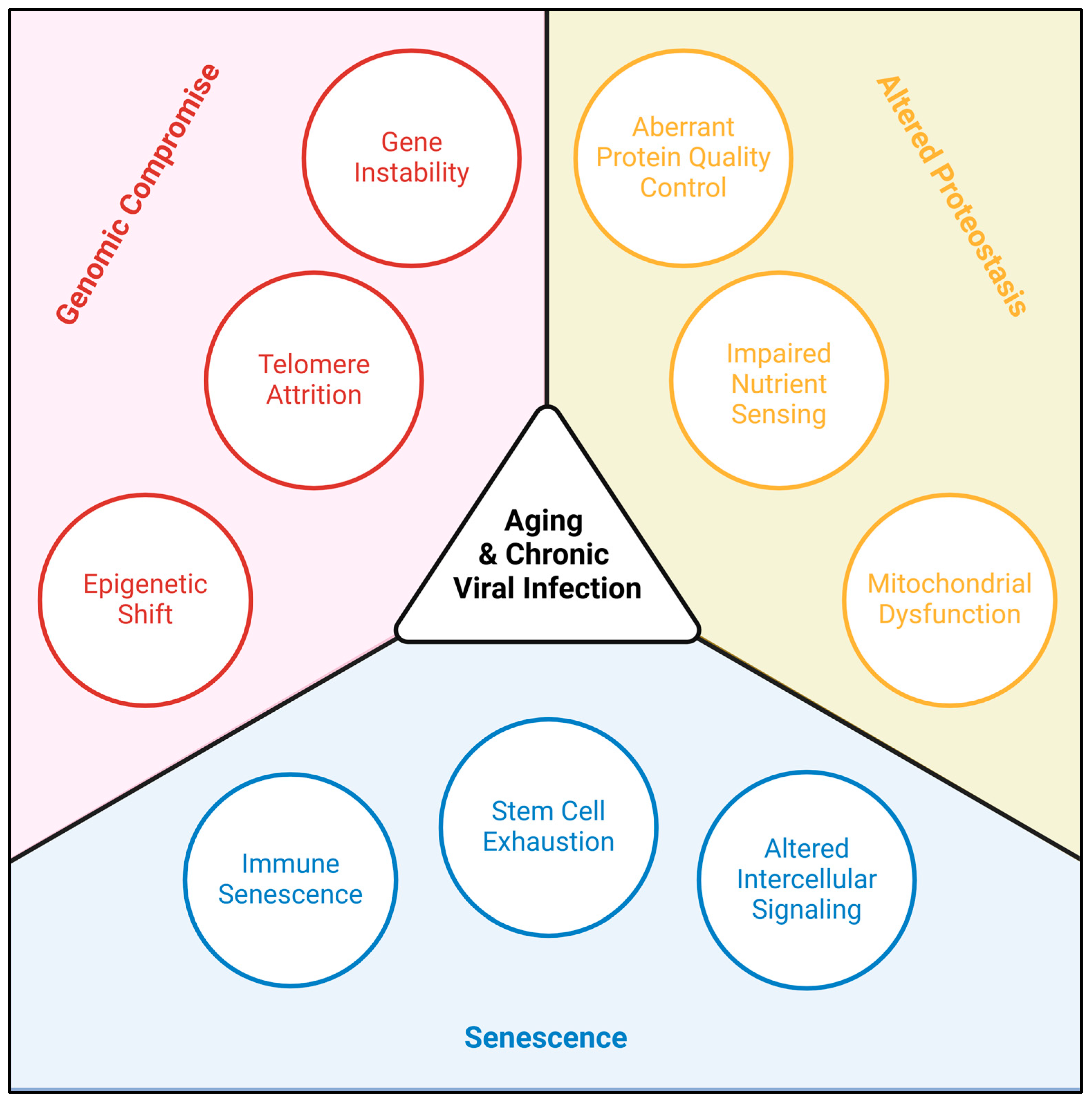
:1. Introduction
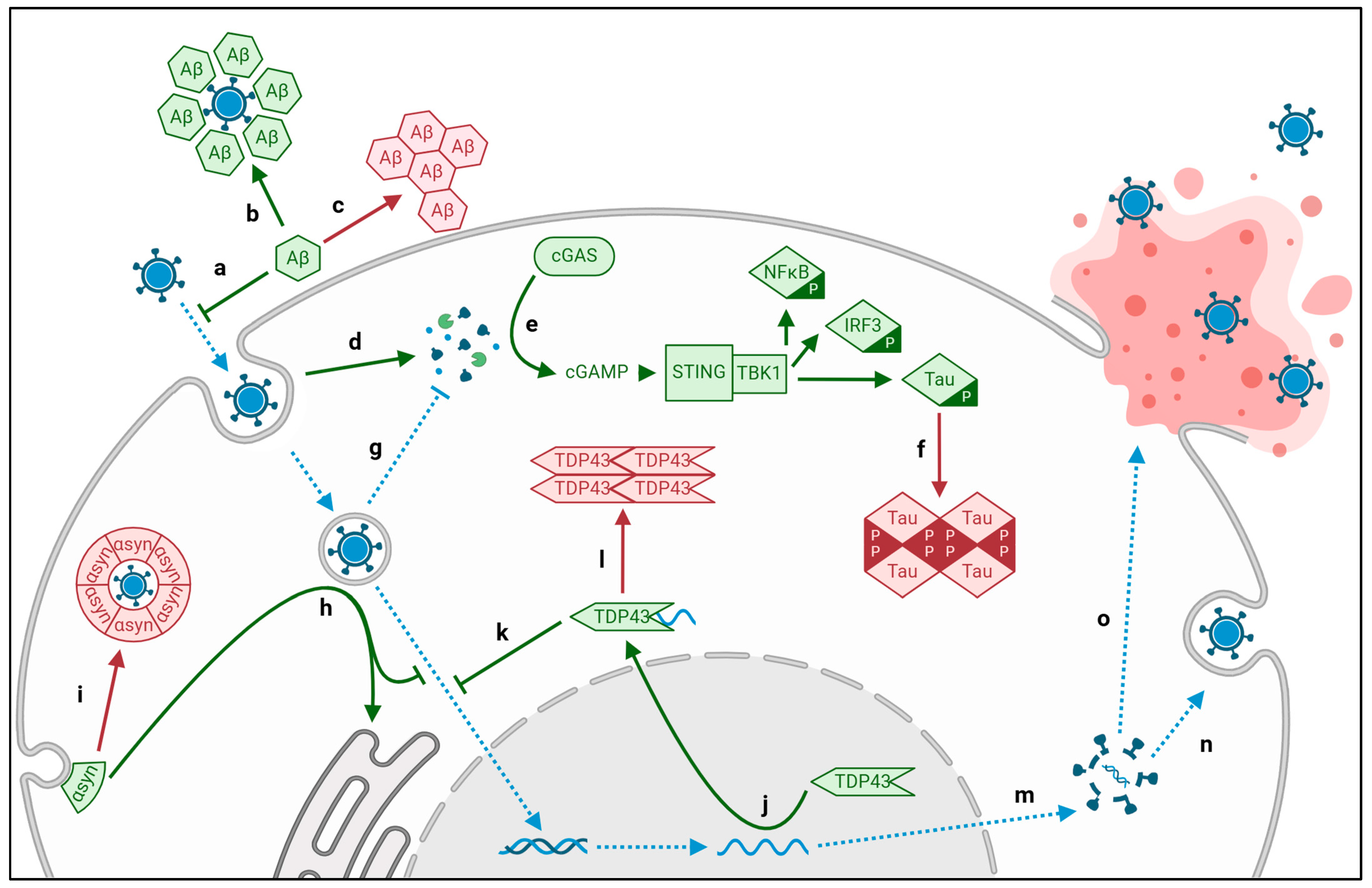
2. Altered Proteostasis
2.1. Viral Aggregation of Intracellular Tau and Extracellular Amyloid-β
2.2. Viral Aggregation of TDP43
2.3. Viral Aggregation of α-Synuclein
2.4. Viral Aberration of Protein Quality Control
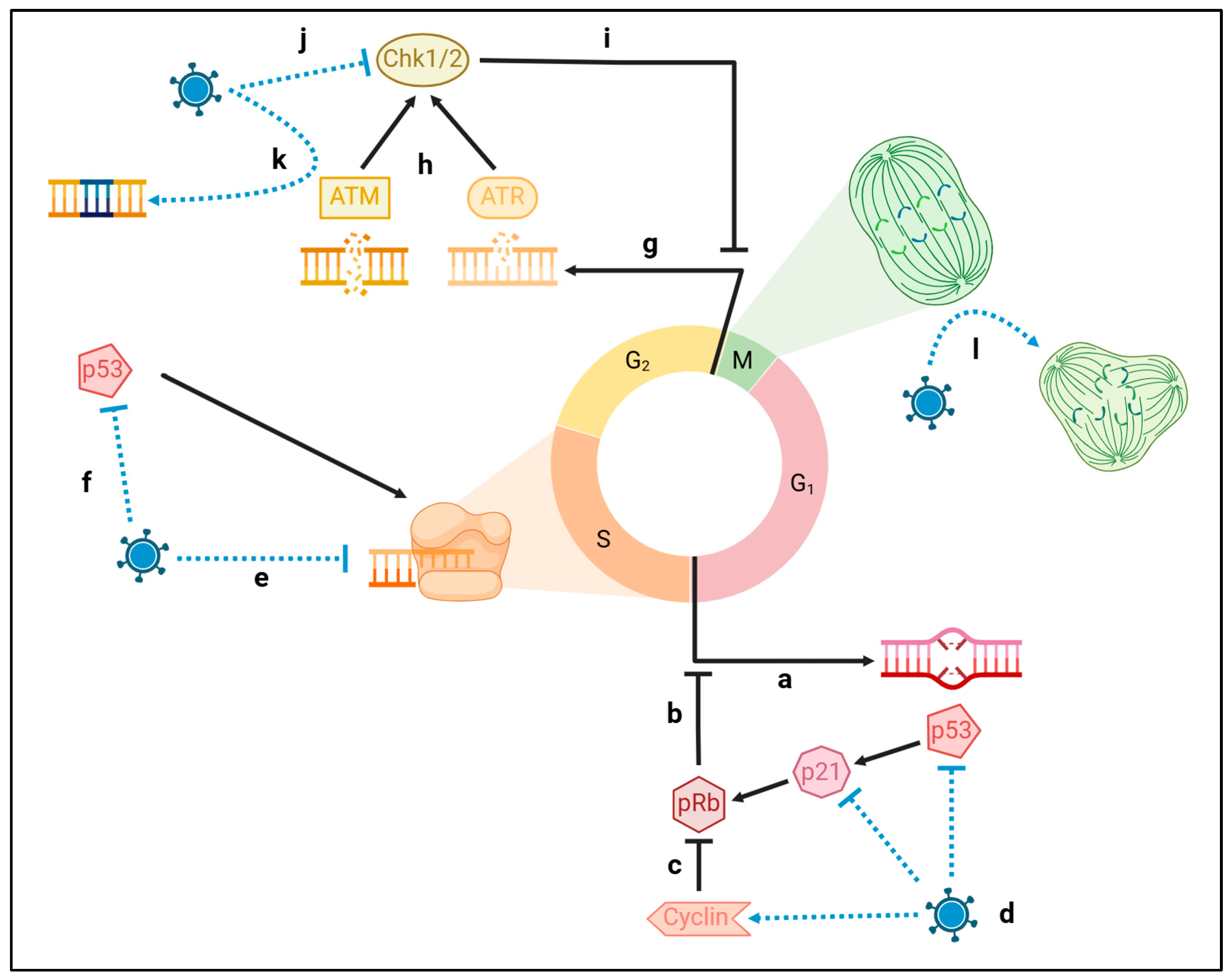
3. Genomic Compromise
3.1. Viral Damage to Host DNA
3.2. Viral Attrition of Telomeres
3.3. Viral Shift of the Epigenome
4. Senescence
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic Acid |
| PQC | Protein quality control |
| ALS | Amyotrophic Lateral Sclerosis |
| PD | Parkinson’s Disease |
| AD | Alzheimer’s Disease |
| FTD | Frontotemporal Dementia |
| RNA | Ribonucleic Acid |
| ssRNA | Single-stranded RNA |
| SARS-CoV-2 | Coronavirus |
| CV | Coxsackie Virus |
| JEV | Japanese encephalitis virus |
| WNV | West Nile virus |
| dsDNA | Double-stranded DNA |
| HSV1 | Herpes Simplex Virus 1 |
| VZV | Varicella Zoster Virus (also human herpesvirus 3) |
| EBV | Epstein–Barr Virus (also human herpesvirus 4) |
| CMV | Cytomegalovirus (also human herpesvirus 5) |
| JCV | JC polyomavirus |
| HIV | Human immunodeficiency virus |
| Aβ | Amyloid-β |
| LBD | Lewy body dementia |
| gD/B | Glycoprotein D/B |
| TDP43 | Transactive response DNA-binding Protein 43 |
| TAT | Transactivator of Transcription |
| cGAS | Cyclic GMP-AMP synthase |
| STING | Synthase-stimulator of Interferon Genes |
| NFκB | Nuclear Factor ΚB |
| IRF3 | Interferon Regulatory Factor 3 |
| TBK1 | TANK-binding Kinase 1 |
| VP | Viral protein |
| GAG | Group-associated Antigen |
| ORF | Open Reading Frame |
| VIF | Viral Infectivity Factor |
| HERV-K | Human Endogenous Retrovirus |
| ASRGL1 | Asparaginase and Isoaspartyl Peptidase 1 |
| ER | Endoplasmic reticulum |
| EBNA | Epstein–Barr Virus Nuclear Antigen |
| DDR | DNA damage response |
| TERT | Telomerase Reverse Transcriptase |
| DNMT | DNA Methyltransferase |
| TET | Ten-eleven Translocation Methylcytosine Dioxygenase |
| PD1 | Programmed Death Receptor 1 |
| CTLA4 | Cytotoxic T Lymphocyte-associated Protein 4 |
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus Exposure and Neurodegenerative Disease Risk across National Biobanks. Neuron 2023, 111, 1086–1093.e2. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Feng, L.; Wu, B.; Xia, W.; Xie, P.; Ma, S.; Liu, H.; Meng, M.; Sun, Y. The Association between Varicella Zoster Virus and Dementia: A Systematic Review and Meta-Analysis of Observational Studies. Neurol. Sci. 2024, 45, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.M.; Smiley, B.M.; Smiley, J.A.; Khorsandian, Y.; Kelly, M.; Itzhaki, R.F.; Kaplan, D.L. Repetitive Injury Induces Phenotypes Associated with Alzheimer’s Disease by Reactivating HSV-1 in a Human Brain Tissue Model. Sci. Signal. 2025, 18, eado6430. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.E.; Colombo, G.; Schulz, R.; Siegert, S. Targeting Microglia with Lentivirus and AAV: Recent Advances and Remaining Challenges. Neurosci. Lett. 2019, 707, 134310. [Google Scholar] [CrossRef]
- Kramm, C.M.; Chase, M.; Herrlinger, U.; Jacobs, A.; Pechan, P.A.; Rainov, N.G.; Sena-Esteves, M.; Aghi, M.; Barnett, F.H.; Chiocca, E.A.; et al. Therapeutic Efficiency and Safety of a Second-Generation Replication-Conditional HSV1 Vector for Brain Tumor Gene Therapy. Hum. Gene Ther. 1997, 8, 2057–2068. [Google Scholar] [CrossRef]
- Kelly, M.J.; O’Keeffe, G.W.; Sullivan, A.M. Viral Vector Delivery of Neurotrophic Factors for Parkinson’s Disease Therapy. Expert. Rev. Mol. Med. 2015, 17, e8. [Google Scholar] [CrossRef]
- Skukan, L.; Brezak, M.; Ister, R.; Klimaschewski, L.; Vojta, A.; Zoldoš, V.; Gajović, S. Lentivirus- or AAV-Mediated Gene Therapy Interventions in Ischemic Stroke: A Systematic Review of Preclinical in Vivo Studies. J. Cereb. Blood Flow. Metab. 2022, 42, 219–236. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Hermann, D.; Gresita, A. Genetic Conversion of Proliferative Astroglia into Neurons after Cerebral Ischemia: A New Therapeutic Tool for the Aged Brain? GeroScience 2019, 41, 363–368. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Song, L.-J.; Wang, Q.; Kumar, G.; Yan, Y.-Q.; Ma, C.-G. Astrocytes: A Double-Edged Sword in Neurodegenerative Diseases. Neural Regen. Res. 2021, 16, 1702. [Google Scholar] [CrossRef]
- Yang, J.-H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of Epigenetic Information as a Cause of Mammalian Aging. Cell 2023, 186, 305–326.e27. [Google Scholar] [CrossRef] [PubMed]
- Bruno, F.; Abondio, P.; Bruno, R.; Ceraudo, L.; Paparazzo, E.; Citrigno, L.; Luiselli, D.; Bruni, A.C.; Passarino, G.; Colao, R.; et al. Alzheimer’s Disease as a Viral Disease: Revisiting the Infectious Hypothesis. Ageing Res. Rev. 2023, 91, 102068. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhao, Y.; Ruan, L.; Zhu, L.; Jin, K.; Zhuge, Q.; Su, D.-M.; Zhao, Y. Impact of Aging Immune System on Neurodegeneration and Potential Immunotherapies. Prog. Neurobiol. 2017, 157, 2–28. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Du, Y.; Zhou, L.; Yue, J.; Hu, X.; Liu, Y.; Chen, S.; Lin, X.; Zhang, G.; Xiao, H.; et al. Gut Microbiota Interact With the Brain Through Systemic Chronic Inflammation: Implications on Neuroinflammation, Neurodegeneration, and Aging. Front. Immunol. 2022, 13, 796288. [Google Scholar] [CrossRef]
- Blinkouskaya, Y.; Caçoilo, A.; Gollamudi, T.; Jalalian, S.; Weickenmeier, J. Brain Aging Mechanisms with Mechanical Manifestations. Mech. Ageing Dev. 2021, 200, 111575. [Google Scholar] [CrossRef]
- Andjelkovic, A.V.; Situ, M.; Citalan-Madrid, A.F.; Stamatovic, S.M.; Xiang, J.; Keep, R.F. Blood-Brain Barrier Dysfunction in Normal Aging and Neurodegeneration: Mechanisms, Impact, and Treatments. Stroke 2023, 54, 661–672. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Jung, H.S.; Bae, S.H.; Chelakkot, C.; Hong, S.; Choi, J.-S.; Yim, D.-S.; Oh, Y.-K.; Choi, Y.-L.; Shin, Y.K. Effect of HPV E6/E7 siRNA with Chemotherapeutic Agents on the Regulation of TP53/E2F Dynamic Behavior for Cell Fate Decisions. Neoplasia 2017, 19, 735–749. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Park, H.; Kam, T.-I.; Dawson, V.L.; Dawson, T.M. α-Synuclein Pathology as a Target in Neurodegenerative Diseases. Nat. Rev. Neurol. 2025, 21, 32–47. [Google Scholar] [CrossRef]
- Koike, Y. Molecular Mechanisms Linking Loss of TDP-43 Function to Amyotrophic Lateral Sclerosis/Frontotemporal Dementia-Related Genes. Neurosci. Res. 2024, 208, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley, P. Fields Virology (Knipe, Fields Virology)-2 Volume Set, 6th ed.; LWW: Philadelphia, PA, USA, 2013; ISBN 978-1-4511-0563-6. [Google Scholar]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The Viral Protein Corona Directs Viral Pathogenesis and Amyloid Aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef] [PubMed]
- Michiels, E.; Rousseau, F.; Schymkowitz, J. Mechanisms and Therapeutic Potential of Interactions between Human Amyloids and Viruses. Cell Mol. Life Sci. 2020, 78, 2485–2501. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Tarenflurbil Phase 3 Study Group Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients with Mild Alzheimer Disease: A Randomized Controlled Trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The Antimicrobial Protection Hypothesis of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Hammarström, P.; Nyström, S. Viruses and Amyloids—A Vicious Liaison. Prion 2023, 17, 82–104. [Google Scholar] [CrossRef]
- Wang, H.-C.; Zhang, Q.-X.; Zhao, J.; Wei, N.-N. Molecular Docking and Molecular Dynamics Simulations Studies on the Protective and Pathogenic Roles of the Amyloid-β Peptide between Herpesvirus Infection and Alzheimer’s Disease. J. Mol. Graph. Model. 2022, 113, 108143. [Google Scholar] [CrossRef]
- Idrees, D.; Kumar, V. SARS-CoV-2 Spike Protein Interactions with Amyloidogenic Proteins: Potential Clues to Neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef]
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and Necroptosis-Associated Proteins Form Heteromeric Amyloid Assemblies. EMBO Rep. 2019, 20, e46518. [Google Scholar] [CrossRef]
- Hategan, A.; Bianchet, M.A.; Steiner, J.; Karnaukhova, E.; Masliah, E.; Fields, A.; Lee, M.-H.; Dickens, A.M.; Haughey, N.; Dimitriadis, E.K.; et al. HIV Tat Protein and Amyloid-β Peptide Form Multifibrillar Structures That Cause Neurotoxicity. Nat. Struct. Mol. Biol. 2017, 24, 379–386. [Google Scholar] [CrossRef]
- Feng, Q.; Hong, Y.; Pradeep Nidamanuri, N.; Yang, C.; Li, Q.; Dong, M. Identification and Nanomechanical Characterization of the HIV Tat-Amyloid β Peptide Multifibrillar Structures. Chem. A Eur. J. 2020, 26, 9449–9453. [Google Scholar] [CrossRef]
- Zhang, K.; Huang, Q.; Li, X.; Zhao, Z.; Hong, C.; Sun, Z.; Deng, B.; Li, C.; Zhang, J.; Wang, S. The cGAS-STING Pathway in Viral Infections: A Promising Link between Inflammation, Oxidative Stress and Autophagy. Front. Immunol. 2024, 15, 1352479. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Hyde, V.R.; Zhou, C.; Fernandez, J.R.; Chatterjee, K.; Ramakrishna, P.; Lin, A.; Fisher, G.W.; Çeliker, O.T.; Caldwell, J.; Bender, O.; et al. Anti-Herpetic Tau Preserves Neurons via the cGAS-STING-TBK1 Pathway in Alzheimer’s Disease. Cell Rep. 2024, 44, 115109. [Google Scholar] [CrossRef]
- Lio, C.-W.J.; McDonald, B.; Takahashi, M.; Dhanwani, R.; Sharma, N.; Huang, J.; Pham, E.; Benedict, C.A.; Sharma, S. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J. Virol. 2016, 90, 7789–7797. [Google Scholar] [CrossRef]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep. 2016, 16, 1492–1501. [Google Scholar] [CrossRef]
- Han, L.; Zhuang, M.-W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.-L.; Zhang, X.-J.; Gao, C.; Wang, P.-H. SARS-CoV-2 ORF9b Antagonizes Type I and III Interferons by Targeting Multiple Components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING Signaling Pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef]
- Marques, M.; Ferreira, A.R.; Ribeiro, D. The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses 2018, 10, 514. [Google Scholar] [CrossRef]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe 2018, 24, 69–80.e4. [Google Scholar] [CrossRef]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein Pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J. Virol. 2018, 92, e01774-17. [Google Scholar] [CrossRef]
- Aguirre, S.; Fernandez-Sesma, A. Collateral Damage during Dengue Virus Infection: Making Sense of DNA by cGAS. J. Virol. 2017, 91, e01081-16. [Google Scholar] [CrossRef] [PubMed]
- Sumner, R.P.; Blest, H.; Lin, M.; Maluquer de Motes, C.; Towers, G.J. HIV-1 with Gag Processing Defects Activates cGAS Sensing. Retrovirology 2024, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wang, W.; Zhao, X.; Jiang, W.; Shao, Q.; Chen, Z.; Huang, C. The Battle between the Innate Immune cGAS-STING Signaling Pathway and Human Herpesvirus Infection. Front. Immunol. 2023, 14, 1235590. [Google Scholar] [CrossRef]
- Xu, G.; Liu, C.; Zhou, S.; Li, Q.; Feng, Y.; Sun, P.; Feng, H.; Gao, Y.; Zhu, J.; Luo, X.; et al. Viral Tegument Proteins Restrict cGAS-DNA Phase Separation to Mediate Immune Evasion. Mol. Cell 2021, 81, 2823–2837.e9. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Lorenzo-Sánchez, I.; Estévez-Herrera, J.; García-Luis, J.; Trujillo-González, R.; Valenzuela-Fernández, A. TDP-43 Controls HIV-1 Viral Production and Virus Infectiveness. Int. J. Mol. Sci. 2023, 24, 7658. [Google Scholar] [CrossRef]
- Fung, G.; Shi, J.; Deng, H.; Hou, J.; Wang, C.; Hong, A.; Zhang, J.; Jia, W.; Luo, H. Cytoplasmic Translocation, Aggregation, and Cleavage of TDP-43 by Enteroviral Proteases Modulate Viral Pathogenesis. Cell Death Differ. 2015, 22, 2087–2097. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, J.; Li, H.; Zhang, Z.; Ji, Z.; Zhao, L.; Wei, W. Enterovirus D68 Infection Induces TDP-43 Cleavage, Aggregation, and Neurotoxicity. J. Virol. 2023, 97, e0042523. [Google Scholar] [CrossRef]
- Xue, Y.C.; Liu, H.; Mohamud, Y.; Bahreyni, A.; Zhang, J.; Cashman, N.R.; Luo, H. Sublethal Enteroviral Infection Exacerbates Disease Progression in an ALS Mouse Model. J. Neuroinflamm. 2022, 19, 16. [Google Scholar] [CrossRef]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front. Mol. Neurosci. 2018, 11, 63. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Fathi, S.; Rastegar, C.; Simula, E.R.; Doucet-O’Hare, T.; Cheng, Y.H.H.; Abrams, R.P.M.; Pasternack, N.; Malik, N.; Bachani, M.; et al. TDP-43 Proteinopathy in ALS Is Triggered by Loss of ASRGL1 and Associated with HML-2 Expression. Nat. Commun. 2024, 15, 4163. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liao, Z.; Tan, H.; Wang, K.; Feng, C.; Xing, P.; Zhang, X.; Hua, J.; Jiang, P.; Peng, S.; et al. The Association between Cytomegalovirus Infection and Neurodegenerative Diseases: A Prospective Cohort Using UK Biobank Data. eClinicalMedicine 2024, 74, 102757. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.-L.; Wang, X.; Xiang, Y.; Shen, L.-L.; Wang, Q.-H.; Liu, Y.-H.; Jiao, S.-S.; Wang, Y.-R.; Cao, H.-Y.; Yi, X.; et al. The Association between Infectious Burden and Parkinson’s Disease: A Case-Control Study. Park. Relat. Disord. 2015, 21, 877–881. [Google Scholar] [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2016, 90, 2767–2782. [Google Scholar] [CrossRef]
- Hopkins, H.K.; Traverse, E.M.; Barr, K.L. Viral Parkinsonism: An Underdiagnosed Neurological Complication of Dengue Virus Infection. PLoS Negl. Trop. Dis. 2022, 16, e0010118. [Google Scholar] [CrossRef]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Callen, S.; Buch, S.; Sawaya, B.E. HIV-1 Vpr Protein Impairs Lysosome Clearance Causing SNCA/Alpha-Synuclein Accumulation in Neurons. Autophagy 2021, 17, 1768–1782. [Google Scholar] [CrossRef]
- Olari, L.-R.; Liu, S.; Arnold, F.; Kühlwein, J.; Gil Miró, M.; Updahaya, A.R.; Stürzel, C.; Thal, D.R.; Walther, P.; Sparrer, K.M.J.; et al. α-Synuclein Fibrils Enhance HIV-1 Infection of Human T Cells, Macrophages and Microglia. Nat. Commun. 2025, 16, 813. [Google Scholar] [CrossRef]
- Gupta, A.; Bohara, V.S.; Chauhan, A.S.; Mohapatra, A.; Kaur, H.; Sharma, A.; Chaudhary, N.; Kumar, S. Alpha-Synuclein Expression in Neurons Modulates Japanese Encephalitis Virus Infection. J. Virol. 2024, 98, e00418-24. [Google Scholar] [CrossRef]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 2021, 13, 143–150. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394. [Google Scholar] [CrossRef]
- Rahic, Z.; Buratti, E.; Cappelli, S. Reviewing the Potential Links between Viral Infections and TDP-43 Proteinopathies. Int. J. Mol. Sci. 2023, 24, 1581. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef] [PubMed]
- Licht-Murava, A.; Meadows, S.M.; Palaguachi, F.; Song, S.C.; Bram, Y.; Zhou, C.; Jackvony, S.; Schwartz, R.E.; Froemke, R.C.; Orr, A.L.; et al. Astrocytic TDP-43 Dysregulation Impairs Memory by Modulating Antiviral Pathways and Interferon-Inducible Chemokines. Sci. Adv. 2023, 9, eade1282. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of Cellular Proteostasis by H1N1 Influenza A Virus Causes α-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Rajendran, A.; Castañeda, C.A. Protein Quality Control Machinery: Regulators of Condensate Architecture and Functionality. Trends Biochem. Sci. 2025, 50, 106–120. [Google Scholar] [CrossRef]
- Almasy, K.M.; Davies, J.P.; Lisy, S.M.; Tirgar, R.; Tran, S.C.; Plate, L. Small-Molecule Endoplasmic Reticulum Proteostasis Regulator Acts as a Broad-Spectrum Inhibitor of Dengue and Zika Virus Infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2012209118. [Google Scholar] [CrossRef]
- Tang, Q.; Wu, P.; Chen, H.; Li, G. Pleiotropic Roles of the Ubiquitin-Proteasome System during Viral Propagation. Life Sci. 2018, 207, 350–354. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during Viral Infection—A Double-Edged Sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Proulx, J.; Borgmann, K.; Park, I.-W. Role of Virally-Encoded Deubiquitinating Enzymes in Regulation of the Virus Life Cycle. Int. J. Mol. Sci. 2021, 22, 4438. [Google Scholar] [CrossRef]
- Gavilán, E.; Medina-Guzman, R.; Bahatyrevich-Kharitonik, B.; Ruano, D. Protein Quality Control Systems and ER Stress as Key Players in SARS-CoV-2-Induced Neurodegeneration. Cells 2024, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.K.; Saulsbery, H.M.; Corona Velazquez, A.F.; Jackson, W.T. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Zeltzer, S.; Zeltzer, C.A.; Igarashi, S.; Wilson, J.; Donaldson, J.G.; Goodrum, F. Virus Control of Trafficking from Sorting Endosomes. mBio 2018, 9, e00683-18. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017, 27, 800. [Google Scholar] [CrossRef]
- Johnston, B.P.; McCormick, C. Herpesviruses and the Unfolded Protein Response. Viruses 2020, 12, 17. [Google Scholar] [CrossRef]
- Rocchi, A.; Sariyer, I.K.; Berger, J.R. Revisiting JC Virus and Progressive Multifocal Leukoencephalopathy. J. Neurovirol. 2023, 29, 524–537. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The Human Cytomegalovirus Protein pUL38 Suppresses Endoplasmic Reticulum Stress-Mediated Cell Death Independently of Its Ability To Induce mTORC1 Activation. J. Virol. 2011, 85, 9103–9113. [Google Scholar] [CrossRef]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr Virus Nuclear Antigen EBNA-LP Is Essential for Transforming Naïve B Cells, and Facilitates Recruitment of Transcription Factors to the Viral Genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef]
- Li, S.; He, S.; Xue, H.; He, Y. Impact of Endogenous Viral Elements on Glioma Clinical Phenotypes by Inducing OCT4 in the Host. Front. Cell Infect. Microbiol. 2024, 14, 1474492. [Google Scholar] [CrossRef]
- Pan, Z.; Huang, X.; Liu, M.; Jiang, X.; He, G. Research Advances in Chaperone-Mediated Autophagy (CMA) and CMA-Based Protein Degraders. J. Med. Chem. 2025, 68, 2314–2332. [Google Scholar] [CrossRef] [PubMed]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Pezone, A.; Olivieri, F.; Napoli, M.V.; Procopio, A.; Avvedimento, E.V.; Gabrielli, A. Inflammation and DNA Damage: Cause, Effect or Both. Nat. Rev. Rheumatol. 2023, 19, 200–211. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Studstill, C.J.; Mac, M.; Moody, C.A. Interplay between the DNA Damage Response and the Life Cycle of DNA Tumor Viruses. Tumour Virus Res. 2023, 16, 200272. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef]
- Plaza-Jennings, A.L.; Valada, A.; O’Shea, C.; Iskhakova, M.; Hu, B.; Javidfar, B.; Ben Hutta, G.; Lambert, T.Y.; Murray, J.; Kassim, B.; et al. HIV Integration in the Human Brain Is Linked to Microglial Activation and 3D Genome Remodeling. Mol. Cell 2022, 82, 4647–4663.e8. [Google Scholar] [CrossRef]
- Li, G.-H.; Maric, D.; Major, E.O.; Nath, A. Productive HIV Infection in Astrocytes Can Be Established via a Non-Classical Mechanism. AIDS 2020, 34, 963–978. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV Integration Sites Are Linked to Clonal Expansion and Persistence of Infected Cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef]
- Wahl, A.; Al-Harthi, L. HIV Infection of Non-Classical Cells in the Brain. Retrovirology 2023, 20, 1. [Google Scholar] [CrossRef]
- Tang, D.; Li, B.; Xu, T.; Hu, R.; Tan, D.; Song, X.; Jia, P.; Zhao, Z. VISDB: A Manually Curated Database of Viral Integration Sites in the Human Genome. Nucleic Acids Res. 2020, 48, D633–D641. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, W.-L.; Zhu, Q.; Zhang, S.; Yao, Y.; Xiang, T.; Feng, Q.-S.; Zhang, Z.; Peng, R.-J.; Jia, W.-H.; et al. Genome-Wide Profiling of Epstein-Barr Virus Integration by Targeted Sequencing in Epstein-Barr Virus Associated Malignancies. Theranostics 2019, 9, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Janjetovic, S.; Hinke, J.; Balachandran, S.; Akyüz, N.; Behrmann, P.; Bokemeyer, C.; Dierlamm, J.; Murga Penas, E.M. Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines. Viruses 2022, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.S.Z.; Abbasi, A.; Kim, D.H.; Lippman, S.M.; Alexandrov, L.B.; Cleveland, D.W. Chromosomal Fragile Site Breakage by EBV-Encoded EBNA1 at Clustered Repeats. Nature 2023, 616, 504–509. [Google Scholar] [CrossRef]
- Lai, M.; Zimmerman, E.S.; Planelles, V.; Chen, J. Activation of the ATR Pathway by Human Immunodeficiency Virus Type 1 Vpr Involves Its Direct Binding to Chromatin In Vivo. J. Virol. 2005, 79, 15443–15451. [Google Scholar] [CrossRef]
- González, M.E. The HIV-1 Vpr Protein: A Multifaceted Target for Therapeutic Intervention. Int. J. Mol. Sci. 2017, 18, 126. [Google Scholar] [CrossRef]
- Gioia, U.; Tavella, S.; Martínez-Orellana, P.; Cicio, G.; Colliva, A.; Ceccon, M.; Cabrini, M.; Henriques, A.C.; Fumagalli, V.; Paldino, A.; et al. SARS-CoV-2 Infection Induces DNA Damage, through CHK1 Degradation and Impaired 53BP1 Recruitment, and Cellular Senescence. Nat. Cell Biol. 2023, 25, 550–564. [Google Scholar] [CrossRef]
- Li, R.; Liao, G.; Nirujogi, R.S.; Pinto, S.M.; Shaw, P.G.; Huang, T.-C.; Wan, J.; Qian, J.; Gowda, H.; Wu, X.; et al. Phosphoproteomic Profiling Reveals Epstein-Barr Virus Protein Kinase Integration of DNA Damage Response and Mitotic Signaling. PLOS Pathog. 2015, 11, e1005346. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting P53 Pathways: Mechanisms, Structures and Advances in Therapy. Sig Transduct. Target. Ther. 2023, 8, 1–35. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.-H.; Schlosser, Y.T.; Kratz, A.-S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein–Barr Virus Particles Induce Centrosome Amplification and Chromosomal Instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef]
- Park, J.-E.; Kim, T.-S.; Zeng, Y.; Mikolaj, M.; Il Ahn, J.; Alam, M.S.; Monnie, C.M.; Shi, V.; Zhou, M.; Chun, T.-W.; et al. Centrosome Amplification and Aneuploidy Driven by the HIV-1-Induced Vpr•VprBP•Plk4 Complex in CD4+ T Cells. Nat. Commun. 2024, 15, 2017. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Murakami, M.; Kumar, P.; Bajaj, B.; Sims, K.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Augments Mdm2-Mediated P53 Ubiquitination and Degradation by Deubiquitinating Mdm2. J. Virol. 2009, 83, 4652–4669. [Google Scholar] [CrossRef] [PubMed]
- Cheerathodi, M.R.; Meckes, D.G. The Epstein-Barr Virus LMP1 Interactome: Biological Implications and Therapeutic Targets. Future Virol. 2018, 13, 863–887. [Google Scholar] [CrossRef]
- Gonzalez, H.; Bolgert, F.; Camporo, P.; Leblond, V. Progressive Multifocal Leukoencephalitis (PML) in Three Patients Treated with Standard-Dose Fludarabine (FAMP). Hematol. Cell Ther. 1999, 41, 183–186. [Google Scholar] [CrossRef]
- Ahye, N.; Bellizzi, A.; May, D.; Wollebo, H.S. The Role of the JC Virus in Central Nervous System Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 6236. [Google Scholar] [CrossRef]
- Marongiu, L.; Allgayer, H. Viruses in Colorectal Cancer. Mol. Oncol. 2022, 16, 1423–1450. [Google Scholar] [CrossRef]
- Zheng, H.-C.; Xue, H.; Zhang, C.-Y. The Oncogenic Roles of JC Polyomavirus in Cancer. Front. Oncol. 2022, 12, 976577. [Google Scholar] [CrossRef]
- Wiebusch, L.; Asmar, J.; Uecker, R.; Hagemeier, C. Human Cytomegalovirus Immediate-Early Protein 2 (IE2)-Mediated Activation of Cyclin E Is Cell-Cycle-Independent and Forces S-Phase Entry in IE2-Arrested Cells. J. Gen. Virol. 2003, 84, 51–60. [Google Scholar] [CrossRef]
- Bogdanow, B.; Phan, Q.V.; Wiebusch, L. Emerging Mechanisms of G1/S Cell Cycle Control by Human and Mouse Cytomegaloviruses. mBio 2021, 12, e02934-21. [Google Scholar] [CrossRef]
- Prichard, M.N.; Sztul, E.; Daily, S.L.; Perry, A.L.; Frederick, S.L.; Gill, R.B.; Hartline, C.B.; Streblow, D.N.; Varnum, S.M.; Smith, R.D.; et al. Human Cytomegalovirus UL97 Kinase Activity Is Required for the Hyperphosphorylation of Retinoblastoma Protein and Inhibits the Formation of Nuclear Aggresomes. J. Virol. 2008, 82, 5054–5067. [Google Scholar] [CrossRef]
- Shukla, S.K.; Jha, H.C.; El-Naccache, D.W.; Robertson, E.S. An EBV Recombinant Deleted for Residues 130-159 in EBNA3C Can Deregulate P53/Mdm2 and Cyclin D1/CDK6 Which Results in Apoptosis and Reduced Cell Proliferation. Oncotarget 2016, 7, 18116–18134. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus Encoded LMP1 Regulates Cyclin D1 Promoter Activity by Nuclear EGFR and STAT3 in CNE1 Cells. J. Exp. Clin. Cancer Res. 2013, 32, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Nakajima, R.; Shirasawa, M.; Fikriyanti, M.; Zhao, L.; Iwanaga, R.; Bradford, A.P.; Kurayoshi, K.; Araki, K.; Ohtani, K. Expanding Roles of the E2F-RB-P53 Pathway in Tumor Suppression. Biology 2023, 12, 1511. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, Health, and Hallmarks of Aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, Z.; Tutton, S.; Lieberman, P.M. The Telomeric Response to Viral Infection. Viruses 2017, 9, 218. [Google Scholar] [CrossRef]
- Nassour, J.; Przetocka, S.; Karlseder, J. Telomeres as Hotspots for Innate Immunity and Inflammation. DNA Repair 2024, 133, 103591. [Google Scholar] [CrossRef]
- Montano, M.; Oursler, K.K.; Xu, K.; Sun, Y.V.; Marconi, V.C. Biological Ageing with HIV Infection: Evaluating the Geroscience Hypothesis. Lancet Healthy Longev. 2022, 3, e194–e205. [Google Scholar] [CrossRef]
- Dowd, J.B.; Bosch, J.A.; Steptoe, A.; Jayabalasingham, B.; Lin, J.; Yolken, R.; Aiello, A.E. Persistent Herpesvirus Infections and Telomere Attrition Over 3 Years in the Whitehall II Cohort. J. Infect. Dis. 2017, 216, 565–572. [Google Scholar] [CrossRef]
- Noppert, G.A.; Feinstein, L.; Dowd, J.B.; Stebbins, R.C.; Zang, E.; Needham, B.L.; Meier, H.C.S.; Simanek, A.; Aiello, A.E. Pathogen Burden and Leukocyte Telomere Length in the United States. Immun. Ageing 2020, 17, 36. [Google Scholar] [CrossRef]
- Virseda-Berdices, A.; Behar-Lagares, R.; Martínez-González, O.; Blancas, R.; Bueno-Bustos, S.; Brochado-Kith, O.; Manteiga, E.; Mallol Poyato, M.J.; López Matamala, B.; Martín Parra, C.; et al. Longer ICU Stay and Invasive Mechanical Ventilation Accelerate Telomere Shortening in COVID-19 Patients 1 Year after Recovery. Crit. Care 2024, 28, 267. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. A Loopy View of Telomere Evolution. Front. Genet. 2015, 6, 321. [Google Scholar] [CrossRef]
- Fischer, N. Infection-Induced Epigenetic Changes and Their Impact on the Pathogenesis of Diseases. Semin. Immunopathol. 2020, 42, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Silmon de Monerri, N.C.; Kim, K. Pathogens Hijack the Epigenome: A New Twist on Host-Pathogen Interactions. Am. J. Pathol. 2014, 184, 897–911. [Google Scholar] [CrossRef]
- Pei, Y.; Robertson, E.S. The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection. Viruses 2020, 12, 1377. [Google Scholar] [CrossRef]
- Christiansen, L.; Lenart, A.; Tan, Q.; Vaupel, J.W.; Aviv, A.; McGue, M.; Christensen, K. DNA Methylation Age Is Associated with Mortality in a Longitudinal Danish Twin Study. Aging Cell 2016, 15, 149–154. [Google Scholar] [CrossRef]
- Bose, D.; Robertson, E.S. Chapter 108—Viruses, Cell Transformation, and Cancer. In Molecular Medical Microbiology, 3rd ed.; Tang, Y.-W., Hindiyeh, M.Y., Liu, D., Sails, A., Spearman, P., Zhang, J.-R., Eds.; Academic Press: Cambridge, MA, USA, 2024; pp. 2209–2225. ISBN 978-0-12-818619-0. [Google Scholar]
- Chen, M.; Lejeune, S.; Zhou, X.; Nadeau, K. Chapter 5—Basic Genetics and Epigenetics for the Immunologist and Allergist. In Allergic and Immunologic Diseases; Chang, C., Ed.; Academic Press: Cambridge, MA, USA, 2022; pp. 119–143. ISBN 978-0-323-95061-9. [Google Scholar]
- Rowles, D.L.; Tsai, Y.-C.; Greco, T.M.; Lin, A.E.; Li, M.; Yeh, J.; Cristea, I.M. DNA Methyltransferase DNMT3A Associates with Viral Proteins and Impacts HSV-1 Infection. PROTEOMICS 2015, 15, 1968–1982. [Google Scholar] [CrossRef]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr Virus Latency in B Cells Leads to Epigenetic Repression and CpG Methylation of the Tumour Suppressor Gene Bim. PLOS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X.; et al. Targeting the Signaling in Epstein–Barr Virus-Associated Diseases: Mechanism, Regulation, and Clinical Study. Sig Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar] [CrossRef]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front. Oncol. 2021, 11, 629780. [Google Scholar] [CrossRef]
- Nehme, Z.; Pasquereau, S.; Herbein, G. Control of Viral Infections by Epigenetic-Targeted Therapy. Clin. Epigenet. 2019, 11, 55. [Google Scholar] [CrossRef]
- Kananen, L.; Nevalainen, T.; Jylhävä, J.; Marttila, S.; Hervonen, A.; Jylhä, M.; Hurme, M. Cytomegalovirus Infection Accelerates Epigenetic Aging. Exp. Gerontol. 2015, 72, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Li, Y.; Zhou, J.; Zhang, Y.; Cao, R. Acute Enterovirus Infections Significantly Alter Host Cellular DNA Methylation Status. Infect. Genet. Evol. 2020, 80, 104190. [Google Scholar] [CrossRef] [PubMed]
- Kuss-Duerkop, S.K.; Westrich, J.A.; Pyeon, D. DNA Tumor Virus Regulation of Host DNA Methylation and Its Implications for Immune Evasion and Oncogenesis. Viruses 2018, 10, 82. [Google Scholar] [CrossRef]
- Balnis, J.; Madrid, A.; Hogan, K.J.; Drake, L.A.; Adhikari, A.; Vancavage, R.; Singer, H.A.; Alisch, R.S.; Jaitovich, A. Persistent Blood DNA Methylation Changes One Year after SARS-CoV-2 Infection. Clin. Epigenet. 2022, 14, 94. [Google Scholar] [CrossRef]
- Esteban-Cantos, A.; Rodríguez-Centeno, J.; Silla, J.C.; Barruz, P.; Sánchez-Cabo, F.; Saiz-Medrano, G.; Nevado, J.; Mena-Garay, B.; Jiménez-González, M.; de Miguel, R.; et al. Effect of HIV Infection and Antiretroviral Therapy Initiation on Genome-Wide DNA Methylation Patterns. eBioMedicine 2023, 88, 104434. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Barthel, S.; Wang, P.; Fikrig, E. Dengue Virus Capsid Protein Binds Core Histones and Inhibits Nucleosome Formation in Human Liver Cells. PLoS ONE 2011, 6, e24365. [Google Scholar] [CrossRef]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the Histone Demethylase LSD1 Blocks Alpha-Herpesvirus Lytic Replication and Reactivation from Latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef]
- Ambagala, A.P.; Bosma, T.; Ali, M.A.; Poustovoitov, M.; Chen, J.J.; Gershon, M.D.; Adams, P.D.; Cohen, J.I. Varicella-Zoster Virus Immediate-Early 63 Protein Interacts with Human Antisilencing Function 1 Protein and Alters Its Ability to Bind Histones H3.1 and H3.3. J. Virol. 2009, 83, 200–209. [Google Scholar] [CrossRef]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Recruits Histone Deacetylase Activity and Associates with the Corepressors mSin3A and NCoR in Human B-Cell Lines. J. Virol. 2003, 77, 4261–4272. [Google Scholar] [CrossRef]
- Park, J.-J.; Kim, Y.-E.; Pham, H.T.; Kim, E.T.; Chung, Y.-H.; Ahn, J.-H. Functional Interaction of the Human Cytomegalovirus IE2 Protein with Histone Deacetylase 2 in Infected Human Fibroblasts. J. Gen. Virol. 2007, 88, 3214–3223. [Google Scholar] [CrossRef] [PubMed]
- Hackett, B.A.; Dittmar, M.; Segrist, E.; Pittenger, N.; To, J.; Griesman, T.; Gordesky-Gold, B.; Schultz, D.C.; Cherry, S. Sirtuin Inhibitors Are Broadly Antiviral against Arboviruses. mBio 2019, 10, e01446-19. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally Occurring P16(Ink4a)-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Ferguson, F.G.; Wikby, A.; Maxson, P.; Olsson, J.; Johansson, B. Immune Parameters in a Longitudinal Study of a Very Old Population of Swedish People: A Comparison between Survivors and Nonsurvivors. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, B378–B382. [Google Scholar] [CrossRef]
- Hadrup, S.R.; Strindhall, J.; Køllgaard, T.; Seremet, T.; Johansson, B.; Pawelec, G.; thor Straten, P.; Wikby, A. Longitudinal Studies of Clonally Expanded CD8 T Cells Reveal a Repertoire Shrinkage Predicting Mortality and an Increased Number of Dysfunctional Cytomegalovirus-Specific T Cells in the Very Elderly. J. Immunol. 2006, 176, 2645–2653. [Google Scholar] [CrossRef]
- Campos, C.; Pera, A.; Sanchez-Correa, B.; Alonso, C.; Lopez-Fernandez, I.; Morgado, S.; Tarazona, R.; Solana, R. Effect of Age and CMV on NK Cell Subpopulations. Exp. Gerontol. 2014, 54, 130–137. [Google Scholar] [CrossRef]
- Pita-Lopez, M.L.; Gayoso, I.; DelaRosa, O.; Casado, J.G.; Alonso, C.; Muñoz-Gomariz, E.; Tarazona, R.; Solana, R. Effect of Ageing on CMV-Specific CD8 T Cells from CMV Seropositive Healthy Donors. Immun. Ageing 2009, 6, 11. [Google Scholar] [CrossRef]
- Wertheimer, A.M.; Bennett, M.S.; Park, B.; Uhrlaub, J.L.; Martinez, C.; Pulko, V.; Currier, N.L.; Nikolich-Žugich, D.; Kaye, J.; Nikolich-Žugich, J. Aging and Cytomegalovirus Infection Differentially and Jointly Affect Distinct Circulating T Cell Subsets in Humans. J. Immunol. 2014, 192, 2143–2155. [Google Scholar] [CrossRef]
- Lopez-Vergès, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 Defines a Functionally Distinct Population of Mature NK Cells in the Human CD56dimCD16+ NK-Cell Subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Ghamar Talepoor, A.; Doroudchi, M. Immunosenescence in Atherosclerosis: A Role for Chronic Viral Infections. Front. Immunol. 2022, 13, 945016. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A.H. Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals1. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Attaf, M.; Malik, A.; Severinsen, M.C.; Roider, J.; Ogongo, P.; Buus, S.; Ndung’u, T.; Leslie, A.; Kløverpris, H.N.; Matthews, P.C.; et al. Major TCR Repertoire Perturbation by Immunodominant HLA-B*44:03-Restricted CMV-Specific T Cells. Front. Immunol. 2018, 9, 2539. [Google Scholar] [CrossRef]
- Nicoli, F.; Clave, E.; Wanke, K.; von Braun, A.; Bondet, V.; Alanio, C.; Douay, C.; Baque, M.; Lependu, C.; Marconi, P.; et al. Primary Immune Responses Are Negatively Impacted by Persistent Herpesvirus Infections in Older People: Results from an Observational Study on Healthy Subjects and a Vaccination Trial on Subjects Aged More than 70 Years Old. eBioMedicine 2022, 76, 103852. [Google Scholar] [CrossRef]
- Lanfermeijer, J.; de Greef, P.C.; Hendriks, M.; Vos, M.; van Beek, J.; Borghans, J.A.M.; van Baarle, D. Age and CMV-Infection Jointly Affect the EBV-Specific CD8+ T-Cell Repertoire. Front. Aging 2021, 2, 665637. [Google Scholar] [CrossRef]
- Campbell, T.M.; McSharry, B.P.; Steain, M.; Ashhurst, T.M.; Slobedman, B.; Abendroth, A. Varicella Zoster Virus Productively Infects Human Natural Killer Cells and Manipulates Phenotype. PLoS Pathog. 2018, 14, e1006999. [Google Scholar] [CrossRef]
- Gerada, C.; Campbell, T.M.; Kennedy, J.J.; McSharry, B.P.; Steain, M.; Slobedman, B.; Abendroth, A. Manipulation of the Innate Immune Response by Varicella Zoster Virus. Front. Immunol. 2020, 11, 1. [Google Scholar] [CrossRef]
- Levin, M.J. Immune Senescence and Vaccines to Prevent Herpes Zoster in Older Persons. Curr. Opin. Immunol. 2012, 24, 494–500. [Google Scholar] [CrossRef]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and Inflammaging in the Aging Process: Age-Related Diseases or Longevity? Ageing Res. Rev. 2021, 71, 101422. [Google Scholar] [CrossRef]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Nicot, C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses 2017, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Derhovanessian, E.; Maier, A.B.; Hähnel, K.; Zelba, H.; de Craen, A.J.M.; Roelofs, H.; Slagboom, E.P.; Westendorp, R.G.J.; Pawelec, G. Lower Proportion of Naïve Peripheral CD8+ T Cells and an Unopposed Pro-Inflammatory Response to Human Cytomegalovirus Proteins in Vitro Are Associated with Longer Survival in Very Elderly People. Age 2013, 35, 1387–1399. [Google Scholar] [CrossRef]
- Pawelec, G.; McElhaney, J.E.; Aiello, A.E.; Derhovanessian, E. The Impact of CMV Infection on Survival in Older Humans. Curr. Opin. Immunol. 2012, 24, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Goldeck, D.; Derhovanessian, E. Inflammation, Ageing and Chronic Disease. Curr. Opin. Immunol. 2014, 29, 23–28. [Google Scholar] [CrossRef]
- Smithey, M.J.; Li, G.; Venturi, V.; Davenport, M.P.; Nikolich-Žugich, J. Lifelong Persistent Viral Infection Alters the Naive T Cell Pool, Impairing CD8 T Cell Immunity in Late Life. J. Immunol. 2012, 189, 5356–5366. [Google Scholar] [CrossRef]
- Serriari, N.-E.; Gondois-Rey, F.; Guillaume, Y.; Remmerswaal, E.B.M.; Pastor, S.; Messal, N.; Truneh, A.; Hirsch, I.; van Lier, R.A.W.; Olive, D. B and T Lymphocyte Attenuator Is Highly Expressed on CMV-Specific T Cells during Infection and Regulates Their Function. J. Immunol. 2010, 185, 3140–3148. [Google Scholar] [CrossRef]
- Effros, R.B.; Boucher, N.; Porter, V.; Zhu, X.; Spaulding, C.; Walford, R.L.; Kronenberg, M.; Cohen, D.; Schächter, F. Decline in CD28+ T Cells in Centenarians and in Long-Term T Cell Cultures: A Possible Cause for Both in Vivo and in Vitro Immunosenescence. Exp. Gerontol. 1994, 29, 601–609. [Google Scholar] [CrossRef]
- Heath, J.J.; Grant, M.D. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells 2020, 9, 766. [Google Scholar] [CrossRef]
- Yoon, M.; Yang, P.-S.; Jin, M.-N.; Yu, H.T.; Kim, T.-H.; Jang, E.; Uhm, J.-S.; Pak, H.-N.; Lee, M.-H.; Joung, B. Association of Physical Activity Level With Risk of Dementia in a Nationwide Cohort in Korea. JAMA Netw. Open 2021, 4, e2138526. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, P.; Zhong, J.; Wang, J.; Xu, Y.; Huang, H.; Liu, X.; Sun, W. Modifiable Lifestyle Factors Influencing Neurological and Psychiatric Disorders Mediated by Structural Brain Reserve: An Observational and Mendelian Randomization Study. J. Affect. Disord. 2025, 372, 440–450. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Genus | Genome | Capsid | Virus | Associated Diseases |
|---|---|---|---|---|
| Coronavirus | Linear +ssRNA | Enveloped Icosahedral | SARS-CoV-2 | COVID-19 |
| Enteroviruses | Linear +ssRNA | Non-enveloped Icosahedral | Coxsackievirus | Hand-Foot-and-Mouth, Viral Meningitis |
| Echovirus | Viral Meningitis | |||
| Poliovirus | Paralytic Poliomyelitis | |||
| Flaviviruses | Linear +ssRNA | Enveloped Dimeric αhelix | Dengue | Breakbone Fever |
| Japanese Encephalitis | Viral Encephalitis | |||
| West Nile | Viral Encephalitis | |||
| Herpesviruses | Linear dsDNA | Enveloped Icosahedral | Herpes Simplex 1 | Cold Sores, Viral Encephalitis |
| Varicella Zoster | Chicken Pox, Shingles | |||
| Epstein–Barr | Cancer (Lymphoma, Leukemia, Nasopharyngeal Carcinoma), Infectious Mononucleosis, Multiple Sclerosis | |||
| Cytomegalovirus | Congenital Birth Defects, Viral Encephalitis | |||
| Polyomaviruses | Circular dsDNA | Non-enveloped Icosahedral | JC | Progressive Multifocal Leukoencephalopathy, Cancer (Glioblastoma, Colorectal Carcinoma) |
| Lentiviruses | Linear +ssRNA | Enveloped Cone-shaped | Human immunodeficiency virus | Acquired Immunodeficiency Syndrome, HIV-associated Neurocognitive Disorder |
| Disease | Proteinopathy | Pathway | Virus | Protein | Citations |
|---|---|---|---|---|---|
| Alzheimer’s Dementia | Extracellular Aβ Plaques | Amyloidogenesis | CMV | M45 | [23] |
| HIV | TAT | [24,25] | |||
| HSVI | gD | [26] | |||
| SARS-CoV-2 | S-protein | [27] | |||
| Intracellular Tau Tangles | cGAS-STING | CMV | pUL31 * pUL83 * | [28,29] | |
| Dengue | NS2B, NS3, NS2B3 | [30] | |||
| HIV | GAG * | [31] | |||
| HSV1 | ICP27 VP11 * | [32,33] | |||
| SARS-CoV-2 | S-protein | [34] | |||
| VZV | ORF9 * | [35] | |||
| Amyotrophic Lateral Sclerosis & Frontotemporal Dementia | Cytosolic TDP43 Aggregates | RNA Translocation | CV | 2A, 2C | [36,37,38,39,40] |
| Echovirus | |||||
| Poliovirus | |||||
| HIV | GAG VIF | [36] | |||
| HERV-K | ASRGL1 * | [41] | |||
| SARS-CoV-2 | S-protein | [27] | |||
| Parkinson’s Disease & Lewy Body Dementia | Lewy body Aggregates | Endoplasmic Reticulum Sequestration | CMV | Envelope | [42] |
| EBV | [43,44] | ||||
| Dengue | [45] | ||||
| HIV | [46,47] | ||||
| JEV | [48] | ||||
| WNV | [44] | ||||
| SARS-CoV-2 | N-protein S-protein | [49,50] |
| Target | Virus | Protein | Mechanism | Citations |
|---|---|---|---|---|
| ATM/ATR | EBV | EBNA3c | Evasion of ATM via p53 degradation | [99] |
| LMP1 | Transcriptional downregulation of ATM | [100] | ||
| HIV | VPR | Chromatin binding activates ATR | [96] | |
| Chk1/2 | EBV | EBNA3a | Inactivation by direct binding | [101] |
| HIV | VPR | Inactivation by phosphorylation | [102] | |
| SARS-CoV-2 | ORF6 NSP13 | Proteolysis Autophagy-mediated degradation | [98] | |
| p53 | EBV | EBNA3c | Ubiquitin-directed degradation | [103] |
| JCV | LTAg | Inactivation by direct binding | [104,105,106] | |
| pRb | CMV | IE2 | Inactivating phosphorylation via Cyclin-E1 | [107] |
| pp71 | Ubiquitin-directed degradation | [108] | ||
| pUL97 | Inactivation by phosphorylation | [109] | ||
| EBV | EBNA3 | Inactivation by direct binding Inactivating phosphorylation via Cyclin-D1 | [110] | |
| LMP1 | Inactivating phosphorylation via Cyclin-D1 | [111] | ||
| JCV | LTAg | Inactivation by direct binding | [104,105,106] |
| Virus | Immune Changes | Citations |
|---|---|---|
| CMV | Decrease in percentage of CD16− NK cells | [151] |
| Decrease in percentage of CD16+/CD56bright NK cells | ||
| Increased CD16+/CD56− subset | [152] | |
| Increased CD8+ T-cells with high CD244 expression | [153] | |
| Increased CD4+ and CD8+ effector memory cells | [154] | |
| Exhaustion of peripheral T-cell compartments | [155] | |
| Accumulation of terminally differentiated, apoptosis-resistant, CMV-specific CD8+ lymphocytes | [156,157] | |
| Reduced diversity of TCR repertoire | [158] | |
| EBV | Increase in differentiated phenotype markers (i.e., KLRG1) | [159] |
| Increase in terminally differentiated T-cells | ||
| Reduced diversity of TCR repertoire | ||
| VZV | Increased population of CD57+, terminally differentiated NK cells | [160] |
| Impaired Type I IFN pathway | [161] | |
| Impaired production of pro-inflammatory cytokines | ||
| Reduced frequency of VZV-specific memory T cells | [162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocchi, A.; Wollebo, H.S.; Khalili, K. Neurotropic Viruses as Acute and Insidious Drivers of Aging. Biomolecules 2025, 15, 514. https://doi.org/10.3390/biom15040514
Rocchi A, Wollebo HS, Khalili K. Neurotropic Viruses as Acute and Insidious Drivers of Aging. Biomolecules. 2025; 15(4):514. https://doi.org/10.3390/biom15040514
Chicago/Turabian StyleRocchi, Angela, Hassen S. Wollebo, and Kamel Khalili. 2025. "Neurotropic Viruses as Acute and Insidious Drivers of Aging" Biomolecules 15, no. 4: 514. https://doi.org/10.3390/biom15040514
APA StyleRocchi, A., Wollebo, H. S., & Khalili, K. (2025). Neurotropic Viruses as Acute and Insidious Drivers of Aging. Biomolecules, 15(4), 514. https://doi.org/10.3390/biom15040514