
Oxidative Stress in Huntington’s Disease

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
Abstract
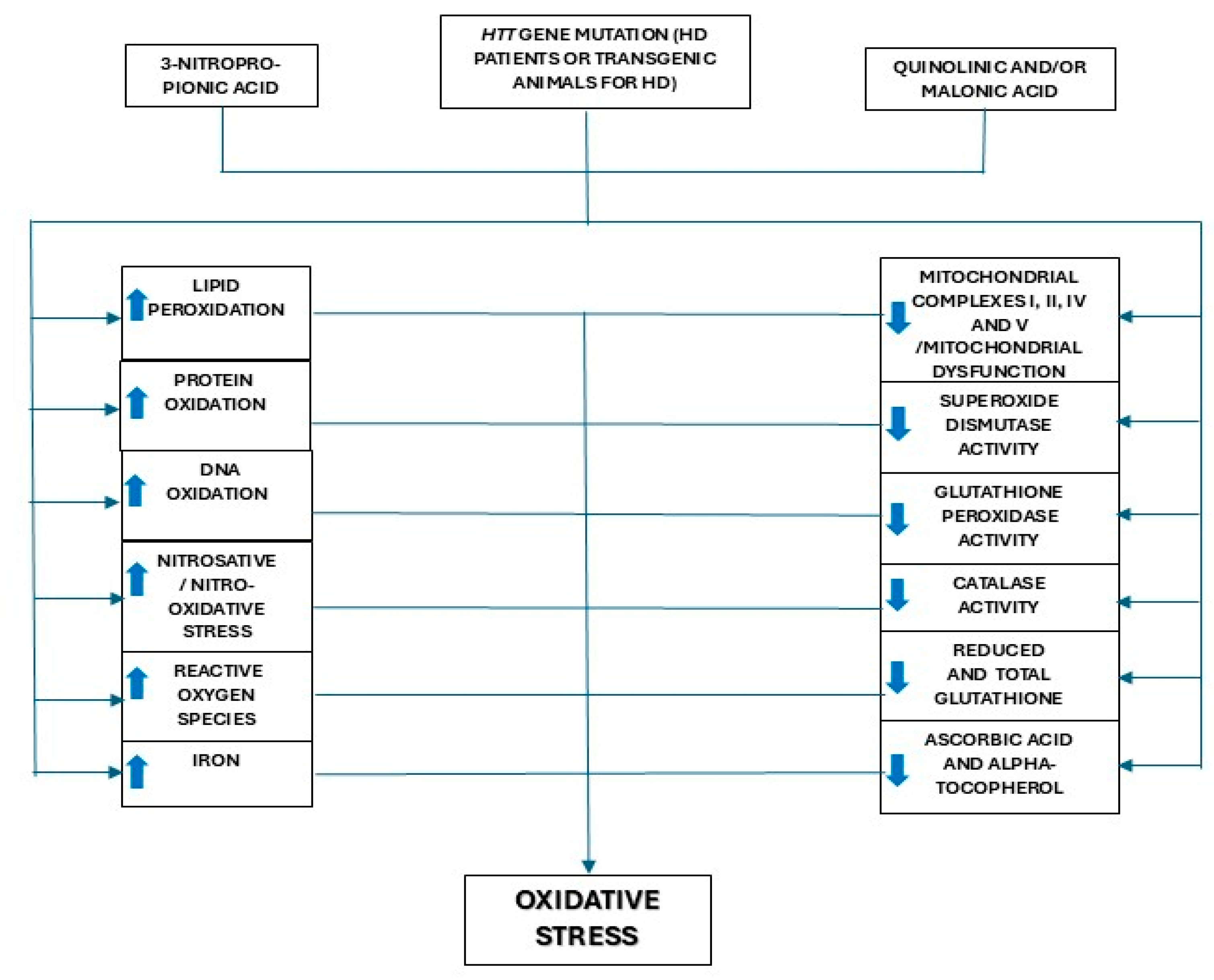
:1. Introduction
2. Oxidative Stress Markers in Patients with Huntington’s Disease
2.1. Oxidative Stress Markers in the Brain
2.2. Oxidative Stress Markers in Plasma/Serum
2.3. Oxidative Stress Markers in Blood Cells and Fibroblasts
2.4. Oxidative Stress Markers in Other Body Fluids
3. Genetic Variants of Genes Related to Oxidative Stress in Patients with Huntington’s Disease
4. Data from Experimental Models of Huntington’s Disease
4.1. Lipid Peroxidation Markers
4.2. Protein Oxidation Markers
4.3. DNA Oxidation Markers
4.4. Nitrosative and Nitrosidative Stress Markers
4.5. Global Oxidative Stress Markers and Trace Metals and Related Proteins
4.6. Mitochondrial Respiratory Chain Complexes
4.7. Proteins, Enzymes, and Vitamins Protective Against Oxidative Stress
- Decrease in glucose-6-phosphate dehydrogenase (G6PD) and 6 phosphogluconate dehydrogenase (6PGD) activities in striatal cell lines of transgenic mice for HD [113].
- Decrease in glyceraldehyde-3-phosphate-dehydrogenase (GADPH) in striatal slices of mice treated with intraperitoneal 3-NPA compared to vehicle controls [107].
- Decrease in monoamine oxidase A (MAO-A) mRNA and activity in cell cultures of mice transgenic for HD compared to wild-type mice [46].
- Non-significant differences in thioredoxin reductase (TRR) activity in brain homogenates of rats treated with intraperitoneal 3-NPA compared to vehicle controls [68].
- Increased expression of peroxiredoxin-1 (Prx-1) in cellular cultures of iPSCs from fibroblasts of YAC128 transgenic mice [123].
- Significant increase in heme oxygenase-1 (HO-1) mRNA and protein expression and HO-1 activity in rats after intrastriatal injection of quinolinic acid compared to vehicle controls [98] and a significant decrease in HO-1 mRNA and protein expression in mice transgenic for HD compared to wild-type mice [91].
- Significant increase in NADPH oxidase (NOX) activity in cellular cultures of a transgenic model of HD in mice after incubation with an organophosphate compound, compared to the wild type [94].
- Significant decrease in vitamin C levels in brain homogenates [65] and plasma [65,105] from rats following intraperitoneal 3-NPA, in brain homogenates of rats after intrastriatal quinolinic acid injection [83,84] compared to vehicle controls, and in transgenic R6/2 mice compared to wild-type mice [128].
5. Discussion and Conclusions
- Increased levels of markers of lipid peroxidation in most studies performed in experimental models and in the plasma of HD patients (studies in the brain are scarce and not conclusive; lipid peroxidation markers were increased in a meta-analysis involving a large number of patients [48]).
- Increased levels of markers of protein and DNA oxidation in most studies performed in experimental models of HD, with inconsistent results for brain samples from HD patients (although a meta-analysis showed increased plasma OH8dG concentrations [48]).
- Increased markers of nitrosative and nitro-oxidative stress in most studies performed in experimental models of HD, while in humans with HD, a study showed no differences in CSF concentration of nitrates + nitrites compared to HCs [49].
- Increased ROS and superoxide anion production and a decrease in global markers of antioxidant status in experimental models of HD.
- Presence of mitochondrial dysfunction and decreased brain complexes I, II, IV, and V of the mitochondrial respiratory chain activities in most of the studies performed using experimental models of HD and a limited number of studies on HD patients.
- Significant decrease in total SOD, GPx, and CAT activities and GSH concentrations in most of the studies using experimental models. Although data obtained in studies with HD brain samples are not conclusive, a meta-analysis of studies on blood oxidative markers of HD patients compared to HCs showed increased GPx activity, similar SOD activity, and decreased GSH levels [48]
- Significant decrease in brain concentrations of vitamins E and C in experimental models of HD.
6. Future Directions
- Design prospective and multicenter studies with a long-term follow-up period (at least 10 years).
- Ensure the participation of a significant number of patients with a genetic–molecular diagnosis of HD, both symptomatic and presymptomatic, as well as a similar number of healthy individuals, matched by age and sex, who are not carriers of HTT gene mutations.
- The participants of the two study groups (HD patients and HCs) involved in the study should only be included after ruling out situations that could influence the oxidative stress measurement parameters, such as therapy with steroids, diuretics, diphosphonate vitamins, calcium or mineral supplements, or drugs that could affect oxidative stress, obesity, undernutrition, pregnancy, oncologic diseases, acute infectious diseases, liver, kidney, thyroid, or parathyroid disease, a recent history of surgery of traumatisms, and atypical dietary habits (for example, diets consisting exclusively of one type of food, such as vegetables).
- It would be desirable to collect plasma/serum, blood cells, and CSF for the analysis of multiple oxidative stress biomarkers, both in HD patients and in HCs, at the baseline and after 5 and 10 years of follow-up.
- Patients with HD should undergo periodic clinical evaluations every 6 months to evaluate the severity and progression of HD, measured according to the Unified Huntington’s Disease Rating Scale [130].
- A new collection of plasma/serum, CSF, and blood cells should be performed for the analysis of multiple oxidative stress biomarkers at the end of the follow-up period.
- Finally, it would be desirable, in the event of death, to obtain a brain donation from patients with HD and healthy HCs to be able to examine the different parameters of oxidative stress in them.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, F.O. Huntington’s Disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A. Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Tong, X.P. Huntington’s Disease: From Basic Science to Therapeutics. CNS Neurosci. Ther. 2018, 24, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.J.; Rodrigues, F.B.; Duarte, G.S.; Mestre, T.A.; Bachoud-Levi, A.C.; Bentivoglio, A.R.; Burgunder, J.M.; Cardoso, F.; Claassen, D.O.; Landwehrmeyer, G.B.; et al. An MDS Evidence-Based Review on Treatments for Huntington’s Disease. Mov. Disord. 2022, 37, 25–35. [Google Scholar] [CrossRef]
- Pan, L.; Feigin, A. Huntington’s Disease: New Frontiers in Therapeutics. Curr. Neurol. Neurosci. Rep. 2021, 21, 10. [Google Scholar] [CrossRef]
- Alam, Z.I.; Halliwell, B.; Jenner, P. No Evidence for Increased Oxidative Damage to Lipids, Proteins, or DNA in Huntington’s Disease. J. Neurochem. 2000, 75, 840–846. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of Lipid Peroxidation and Mitochondrial Function Improves Neuropathology in Huntington’s Disease Mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef]
- Kreilaus, F.; Spiro, A.S.; McLean, C.A.; Garner, B.; Jenner, A.M. Evidence for Altered Cholesterol Metabolism in Huntington’s Disease Post Mortem Brain Tissue. Neuropathol. Appl. Neurobiol. 2016, 42, 535–546. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and Oxidative Stress Analysis in Human Brain Samples of Huntington Disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative Damage and Metabolic Dysfunction in Huntington’s Disease: Selective Vulnerability of the Basal Ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative Damage to Mitochondrial DNA in Huntington’s Disease Parietal Cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Rodríguez-Colman, M.J.; Tamarit, J.; Ortega, Z.; Lucas, J.J.; Ferrer, I.; Ros, J.; Cabiscol, E. Protein Oxidation in Huntington Disease Affects Energy Production and Vitamin B6 Metabolism. Free Radic. Biol. Med. 2010, 49, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Richards, G.; Messer, J.; Waldvogel, H.J.; Gibbons, H.M.; Dragunow, M.; Faull, R.L.; Saura, J. Up-Regulation of the Isoenzymes MAO-A and MAO-B in the Human Basal Ganglia and Pons in Huntington’s Disease Revealed by Quantitative Enzyme Radioautography. Brain Res. 2011, 1370, 204–214. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in Glutathione Levels in Parkinson’s Disease and Other Neurodegenerative Disorders Affecting Basal Ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Scholefield, M.; Patassini, S.; Xu, J.; Cooper, G.J.S. Widespread Selenium Deficiency in the Brain of Cases with Huntington’s Disease Presents a New Potential Therapeutic Target. EBioMedicine 2023, 97, 104824. [Google Scholar] [CrossRef]
- Bartzokis, G.; Cummings, J.; Perlman, S.; Hance, D.B.; Mintz, J. Increased Basal Ganglia Iron Levels in Huntington Disease. Arch. Neurol. 1999, 56, 569–574. [Google Scholar] [CrossRef]
- Bartzokis, G.; Tishler, T.A. MRI Evaluation of Basal Ganglia Ferritin Iron and Neurotoxicity in Alzheimer’s and Huntington’s Disease. Cell. Mol. Biol. 2000, 46, 821–833. [Google Scholar]
- Vymazal, J.; Klempír, J.; Jech, R.; Zidovská, J.; Syka, M.; Růzicka, E.; Roth, J. MR Relaxometry in Huntington’s Disease: Correlation Between Imaging, Genetic and Clinical Parameters. J. Neurol. Sci. 2007, 263, 20–25. [Google Scholar] [CrossRef]
- Simmons, D.A.; Casale, M.; Alcon, B.; Pham, N.; Narayan, N.; Lynch, G. Ferritin Accumulation in Dystrophic Microglia is an Early Event in the Development of Huntington’s Disease. Glia 2007, 55, 1074–1084. [Google Scholar] [CrossRef]
- Loeffler, D.A.; LeWitt, P.A.; Juneau, P.L.; Sima, P.A.; Nguyen, H.U.; DeMaggio, A.J.; Brickman, C.M.; Brewer, G.J.; Dick, R.D.; Troyer, M.D.; et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 1996, 738, 265–274. [Google Scholar] [CrossRef]
- Lu, Z.; Marks, E.; Chen, J.; Moline, J.; Barrows, L.; Raisbeck, M.; Volitakis, I.; Cherny, R.A.; Chopra, V.; Bush, A.I.; et al. Altered selenium status in Huntington’s disease: Neuroprotection by selenite in the N171-82Q mouse model. Neurobiol. Dis. 2014, 71, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Rodríguez-Colman, M.J.; Vall-Llaura, N.; Vived, C.; Fernández-Nogales, M.; Lucas, J.J.; Ferrer, I.; Cabiscol, E. Impaired PLP-dependent metabolism in brain samples from Huntington disease patients and transgenic R6/1 mice. Metab. Brain Dis. 2016, 31, 579–586. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Drouin-Ouellet, J.; Tirolo, C.; Pulvirenti, A.; Giugno, R.; Testa, N.; Caniglia, S.; Serapide, M.F.; Cisbani, G.; Barker, R.A.; et al. GSK-3β-induced Tau pathology drives hippocampal neuronal cell death in Huntington’s disease: Involvement of astrocyte-neuron interactions. Cell Death Dis. 2016, 7, e2206. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Cheng, M.L.; Tang, H.Y.; Lin, C.Y.; Chen, C.M. Dysregulation of choline metabolism and therapeutic potential of citicoline in Huntington’s disease. Aging Cell 2024, 23, e14302. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Haque, A.; Aboufadel, S.; Snell, C.; Fischer, R.S.; Granger, S.W.; Granger, D.A.; Thomas, E.A. Uric Acid as a Potential Peripheral Biomarker for Disease Features in Huntington’s Patients. Front. Neurosci. 2020, 14, 73. [Google Scholar] [CrossRef]
- Stoy, N.; Mackay, G.M.; Forrest, C.M.; Christofides, J.; Egerton, M.; Stone, T.W.; Darlington, L.G. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J. Neurochem. 2005, 93, 611–623. [Google Scholar] [CrossRef]
- Christofides, J.; Bridel, M.; Egerton, M.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; Darlington, L.G.; Stone, T.W. Blood 5-hydroxytryptamine, 5-hydroxyindoleacetic acid and melatonin levels in patients with either Huntington’s disease or chronic brain injury. J. Neurochem. 2006, 97, 1078–1088. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Peña-Sánchez, M.; Riverón-Forment, G.; Zaldívar-Vaillant, T.; Soto-Lavastida, A.; Borrero-Sánchez, J.; Lara-Fernández, G.; Esteban-Hernández, E.M.; Hernández-Díaz, Z.; González-Quevedo, A.; Fernández-Almirall, I.; et al. Association of status redox with demographic, clinical and imaging parameters in patients with Huntington’s disease. Clin. Biochem. 2015, 48, 1258–1263. [Google Scholar] [CrossRef]
- Olsson, M.G.; Davidsson, S.; Muhammad, Z.D.; Lahiri, N.; Tabrizi, S.J.; Akerstrom, B.; Bjorkqvist, M. Increased levels of hemoglobin and alpha1-microglobulin in Huntington’s disease. Front. Biosci. (Elite Ed.) 2012, 4, 950–957. [Google Scholar] [CrossRef]
- Klepac, N.; Relja, M.; Klepac, R.; Hećimović, S.; Babić, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.; Barrero, F.J.; Morales, B.; Luna, J.D.; Ramirez, M.; Vives, F. Oxidative stress and plasma aminopeptidase activity in Huntington’s disease. J. Neural Transm. 2010, 117, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Túnez, I.; Sánchez-López, F.; Agüera, E.; Fernández-Bolaños, R.; Sánchez, F.M.; Tasset-Cuevas, I. Important role of oxidative stress biomarkers in Huntington’s disease. J. Med. Chem. 2011, 54, 5602–5606. [Google Scholar] [CrossRef] [PubMed]
- Biglan, K.M.; Dorsey, E.R.; Evans, R.V.; Ross, C.A.; Hersch, S.; Shoulson, I.; Matson, W.; Kieburtz, K.; Huntington Study Group Pre-2CARE Investigators. Plasma 8-hydroxy-2′-deoxyguanosine Levels in Huntington Disease and Healthy Controls Treated with Coenzyme Q10. J. Huntingt. Dis. 2012, 1, 65–69. [Google Scholar] [CrossRef]
- Long, J.D.; Matson, W.R.; Juhl, A.R.; Leavitt, B.R.; Paulsen, J.S.; PREDICT-HD Investigators and Coordinators of the Huntington Study Group. 8OHdG as a marker for Huntington disease progression. Neurobiol. Dis. 2012, 46, 625–634. [Google Scholar] [CrossRef]
- Sánchez-López, F.; Tasset, I.; Agüera, E.; Feijóo, M.; Fernández-Bolaños, R.; Sánchez, F.M.; Ruiz, M.C.; Cruz, A.H.; Gascón, F.; Túnez, I. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington’s disease. Neurol. Res. 2012, 34, 721–724. [Google Scholar] [CrossRef]
- Kalliolia, E.; Silajdžić, E.; Nambron, R.; Hill, N.R.; Doshi, A.; Frost, C.; Watt, H.; Hindmarsh, P.; Björkqvist, M.; Warner, T.T. Plasma melatonin is reduced in Huntington’s disease. Mov. Disord. 2014, 29, 1511–1515. [Google Scholar] [CrossRef]
- Squadrone, S.; Brizio, P.; Abete, M.C.; Brusco, A. Trace elements profile in the blood of Huntington’ disease patients. J. Trace Elem. Med. Biol. 2020, 57, 18–20. [Google Scholar] [CrossRef]
- Ciancarelli, I.; De Amicis, D.; Di Massimo, C.; Di Scanno, C.; Pistarini, C.; D’Orazio, N.; Tozzi Ciancarelli, M.G. Peripheral biomarkers of oxidative stress and their limited potential in evaluation of clinical features of Huntington’s patients. Biomarkers 2014, 19, 452–456. [Google Scholar] [CrossRef]
- Cuturic, M.; Abramson, R.K.; Moran, R.R.; Hardin, J.W.; Frank, E.M.; Sellers, A.A. Serum carnitine levels and levocarnitine supplementation in institutionalized Huntington’s disease patients. Neurol. Sci. 2013, 34, 93–98. [Google Scholar] [CrossRef]
- Liu, C.S.; Cheng, W.L.; Kuo, S.J.; Li, J.Y.; Soong, B.W.; Wei, Y.H. Depletion of mitochondrial DNA in leukocytes of patients with poly-Q diseases. J. Neurol. Sci. 2008, 264, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Wu, Y.R.; Chang, K.H. Altered Aconitase 2 Activity in Huntington’s Disease Peripheral Blood Cells and Mouse Model Striatum. Int. J. Mol. Sci. 2017, 18, 2480. [Google Scholar] [CrossRef] [PubMed]
- Zanella, A.; Izzo, C.; Meola, G.; Mariani, M.; Colotti, M.T.; Silani, V.; Pellegata, G.; Scarlato, G. Metabolic impairment and membrane abnormality in red cells from Huntington’s disease. J. Neurol. Sci. 1980, 47, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Jędrak, P.; Mozolewski, P.; Węgrzyn, G.; Więckowski, M.R. Mitochondrial alterations accompanied by oxidative stress conditions in skin fibroblasts of Huntington’s disease patients. Metab. Brain Dis. 2018, 33, 2005–2017. [Google Scholar] [CrossRef]
- del Hoyo, P.; García-Redondo, A.; de Bustos, F.; Molina, J.A.; Sayed, Y.; Alonso-Navarro, H.; Caballero, L.; Arenas, J.; Jiménez-Jiménez, F.J. Oxidative stress in skin fibroblasts cultures of patients with Huntington’s disease. Neurochem. Res. 2006, 31, 1103–1109. [Google Scholar] [CrossRef]
- Ooi, J.; Hayden, M.R.; Pouladi, M.A. Inhibition of Excessive Monoamine Oxidase A/B Activity Protects Against Stress-induced Neuronal Death in Huntington Disease. Mol. Neurobiol. 2015, 52, 1850–1861. [Google Scholar] [CrossRef]
- PerezGrovas-Saltijeral, A.; Ochoa-Morales, A.; Miranda-Duarte, A.; Martínez-Ruano, L.; Jara-Prado, A.; Camacho-Molina, A.; Hidalgo-Bravo, A. Telomere length analysis on leukocytes derived from patients with Huntington Disease. Mech. Ageing Dev. 2020, 185, 111189. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, H.; Shi, X.J.; Cheng, Y. Blood Oxidative Stress Marker Aberrations in Patients with Huntington’s Disease: A Meta-Analysis Study. Oxid. Med. Cell. Longev. 2020, 2020, 9187195. [Google Scholar] [CrossRef]
- Milstien, S.; Sakai, N.; Brew, B.J.; Krieger, C.; Vickers, J.H.; Saito, K.; Heyes, M.P. cerebrospinal fluid nitrite/nitrate levels in neurologic diseases. J. Neurochem. 1994, 63, 1178–1180. [Google Scholar] [CrossRef]
- Weydt, P.; Soyal, S.M.; Gellera, C.; Didonato, S.; Weidinger, C.; Oberkofler, H.; Landwehrmeyer, G.B.; Patsch, W. The gene coding for PGC-1alpha modifies age at onset in Huntington’s Disease. Mol. Neurodegener. 2009, 4, 3. [Google Scholar] [CrossRef]
- Berger, F.; Vaslin, L.; Belin, L.; Asselain, B.; Forlani, S.; Humbert, S.; Durr, A.; Hall, J. The impact of single-nucleotide polymorphisms (SNPs) in OGG1 and XPC on the age at onset of Huntington disease. Mutat. Res. 2013, 755, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Chen, Y.C.; Wu, Y.R.; Lee, W.F.; Chen, C.M. Downregulation of genes involved in metabolism and oxidative stress in the peripheral leukocytes of Huntington’s disease patients. PLoS ONE 2012, 7, e46492. [Google Scholar] [CrossRef]
- Upadhayay, S.; Jamwal, S.; Kumar, P. Animal models of Huntington’s disease and their applicability to novel drug discovery and development. Expert Opin. Drug Discov. 2023, 18, 527–538. [Google Scholar] [CrossRef]
- Morton, A.J. Sleep and Circadian Rhythm Dysfunction in Animal Models of Huntington’s Disease. J. Huntingt. Dis. 2023, 12, 133–148. [Google Scholar] [CrossRef]
- Rana, N.; Kapil, L.; Singh, C.; Singh, A. Modeling Huntington’s disease: An insight on in-vitro and in-vivo models. Behav. Brain Res. 2024, 459, 114757. [Google Scholar] [CrossRef]
- Nittari, G.; Roy, P.; Martinelli, I.; Bellitto, V.; Tomassoni, D.; Traini, E.; Tayebati, S.K.; Amenta, F. Rodent Models of Huntington’s Disease: An Overview. Biomedicines 2023, 11, 3331. [Google Scholar] [CrossRef]
- Han, B.; Liang, W.; Li, X.J.; Li, S.; Yan, S.; Tu, Z. Large animal models for Huntington’s disease research. Zool. Res. 2024, 45, 275–283. [Google Scholar] [CrossRef]
- Túnez, I.; Montilla, P.; Del Carmen Muñoz, M.; Feijóo, M.; Salcedo, M. Protective effect of melatonin on 3-nitropropionic acid-induced oxidative stress in synaptosomes in an animal model of Huntington’s disease. J. Pineal Res. 2004, 37, 252–256. [Google Scholar] [CrossRef]
- Yang, L.; Calingasan, N.Y.; Chen, J.; Ley, J.J.; Becker, D.A.; Beal, M.F. A novel azulenyl nitrone antioxidant protects against MPTP and 3-nitropropionic acid neurotoxicities. Exp. Neurol. 2005, 191, 86–93. [Google Scholar] [CrossRef]
- Túnez, I.; Drucker-Colín, R.; Jimena, I.; Medina, F.J.; Muñoz, M.C.; Peña, J.; Montilla, P. Transcranial magnetic stimulation attenuates cell loss and oxidative damage in the striatum induced in the 3-nitropropionic model of Huntington’s disease. J. Neurochem. 2006, 97, 619–630. [Google Scholar] [CrossRef]
- Kumar, P.; Padi, S.S.; Naidu, P.S.; Kumar, A. Effect of resveratrol on 3-nitropropionic acid-induced biochemical and behavioural changes: Possible neuroprotective mechanisms. Behav. Pharmacol. 2006, 17, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A. Possible role of sertraline against 3-nitropropionic acid induced behavioral, oxidative stress and mitochondrial dysfunctions in rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Mehrotra, A.; Kamboj, S.S. Lycopene prevents 3-nitropropionic acid-induced mitochondrial oxidative stress and dysfunctions in nervous system. Neurochem. Int. 2010, 57, 579–587. [Google Scholar] [CrossRef]
- Tasset, I.; Pontes, A.J.; Hinojosa, A.J.; de la Torre, R.; Túnez, I. Olive oil reduces oxidative damage in a 3-nitropropionic acid-induced Huntington’s disease-like rat model. Nutr. Neurosci. 2011, 14, 106–111. [Google Scholar] [CrossRef]
- Gopinath, K.; Prakash, D.; Sudhandiran, G. Neuroprotective effect of naringin, a dietary flavonoid against 3-nitropropionic acid-induced neuronal apoptosis. Neurochem. Int. 2011, 59, 1066–1073. [Google Scholar] [CrossRef]
- Bhateja, D.K.; Dhull, D.K.; Gill, A.; Sidhu, A.; Sharma, S.; Reddy, B.V.; Padi, S.S. Peroxisome proliferator-activated receptor-α activation attenuates 3-nitropropionic acid induced behavioral and biochemical alterations in rats: Possible neuroprotective mechanisms. Eur. J. Pharmacol. 2012, 674, 33–43. [Google Scholar] [CrossRef]
- Shivasharan, B.D.; Nagakannan, P.; Thippeswamy, B.S.; Veerapur, V.P.; Bansal, P.; Unnikrishnan, M.K. Protective effect of Calendula officinalis Linn. flowers against 3-nitropropionic acid induced experimental Huntington’s disease in rats. Drug Chem. Toxicol. 2013, 36, 466–473. [Google Scholar] [CrossRef]
- Denny Joseph, K.M.; Muralidhara. Enhanced neuroprotective effect of fish oil in combination with quercetin against 3-nitropropionic acid induced oxidative stress in rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 40, 83–92. [Google Scholar] [CrossRef]
- Binawade, Y.; Jagtap, A. Neuroprotective effect of lutein against 3-nitropropionic acid-induced Huntington’s disease-like symptoms: Possible behavioral, biochemical, and cellular alterations. J. Med. Food 2013, 16, 934–943. [Google Scholar] [CrossRef]
- Sandhir, R.; Yadav, A.; Mehrotra, A.; Sunkaria, A.; Singh, A.; Sharma, S. Curcumin nanoparticles attenuate neurochemical and neurobehavioral deficits in experimental model of Huntington’s disease. Neuromol. Med. 2014, 16, 106–118. [Google Scholar] [CrossRef]
- Thangarajan, S.; Deivasigamani, A.; Natarajan, S.S.; Krishnan, P.; Mohanan, S.K. Neuroprotective Activity of L-Theanine on 3-Nitropropionic Acid-Induced Neurotoxicity in Rat Striatum. Int. J. Neurosci. 2014, 124, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Thangarajan, S.; Ramachandran, S.; Krishnamurthy, P. Chrysin Exerts Neuroprotective Effects against 3-Nitropropionic Acid Induced Behavioral Despair-Mitochondrial Dysfunction and Striatal Apoptosis via Upregulating Bcl-2 Gene and Downregulating Bax-Bad Genes in Male Wistar Rats. Biomed. Pharmacother. 2016, 84, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Sharma, B. Pharmacological Benefit of I(1)-Imidazoline Receptors Activation and Nuclear Factor Kappa-B (NF-κB) Modulation in Experimental Huntington’s Disease. Brain Res. Bull. 2014, 102, 57–68. [Google Scholar] [CrossRef]
- Hariharan, A.; Shetty, S.; Shirole, T.; Jagtap, A.G. Potential of Protease Inhibitor in 3-Nitropropionic Acid Induced Huntington’s Disease Like Symptoms: Mitochondrial Dysfunction and Neurodegeneration. Neurotoxicology 2014, 45, 139–148. [Google Scholar] [CrossRef]
- Khan, A.; Jamwal, S.; Bijjem, K.R.; Prakash, A.; Kumar, P. Neuroprotective Effect of Hemeoxygenase-1/Glycogen Synthase Kinase-3β Modulators in 3-Nitropropionic Acid-Induced Neurotoxicity in Rats. Neuroscience 2015, 287, 66–77. [Google Scholar] [CrossRef]
- Courtes, A.A.; Arantes, L.P.; Barcelos, R.P.; da Silva, I.K.; Boligon, A.A.; Athayde, M.L.; Puntel, R.L.; Soares, F.A. Protective Effects of Aqueous Extract of Luehea divaricata against Behavioral and Oxidative Changes Induced by 3-Nitropropionic Acid in Rats. Evid.-Based Complement. Altern. Med. 2015, 2015, 723431. [Google Scholar] [CrossRef]
- Silva-Palacios, A.; Colín-González, A.L.; López-Cervantes, S.P.; Zazueta, C.; Luna-López, A.; Santamaría, A.; Königsberg, M. Tert-Buthylhydroquinone Pre-Conditioning Exerts Dual Effects in Old Female Rats Exposed to 3-Nitropropionic Acid. Redox Biol. 2017, 12, 610–624. [Google Scholar] [CrossRef]
- Badini, F.; Bayrami, A.; Mirshekar, M.A.; Shahraki, S.; Fanaei, H. Levothyroxine Attenuates Behavioral Impairment and Improves Oxidative Stress and Histological Alteration 3-Nitropropionic Acid Induced Experimental Huntington’s Disease in Rats. Behav. Brain Res. 2024, 461, 114864. [Google Scholar] [CrossRef]
- Ryu, J.K.; Choi, H.B.; McLarnon, J.G. Combined Minocycline Plus Pyruvate Treatment Enhances Effects of Each Agent to Inhibit Inflammation, Oxidative Damage, and Neuronal Loss in an Excitotoxic Animal Model of Huntington’s Disease. Neuroscience 2006, 141, 1835–1848. [Google Scholar] [CrossRef]
- Kalonia, H.; Kumar, P.; Nehru, B.; Kumar, A. Neuroprotective Effect of MK-801 against Intra-Striatal Quinolinic Acid Induced Behavioral, Oxidative Stress and Cellular Alterations in Rats. Indian J. Exp. Biol. 2009, 47, 880–892. [Google Scholar]
- Maldonado, P.D.; Molina-Jijón, E.; Villeda-Hernández, J.; Galván-Arzate, S.; Santamaría, A.; Pedraza-Chaverrí, J. NAD(P)H Oxidase Contributes to Neurotoxicity in an Excitotoxic/Prooxidant Model of Huntington’s Disease in Rats: Protective Role of Apocynin. J. Neurosci. Res. 2010, 88, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Kalonia, H.; Kumar, P.; Kumar, A. Targeting Oxidative Stress Attenuates Malonic Acid Induced Huntington Like Behavioral and Mitochondrial Alterations in Rats. Eur. J. Pharmacol. 2010, 634, 46–52. [Google Scholar] [CrossRef]
- Sumathi, T.; Vedagiri, A.; Ramachandran, S.; Purushothaman, B. Quinolinic Acid-Induced Huntington Disease-Like Symptoms Mitigated by Potent Free Radical Scavenger Edaravone—A Pilot Study on Neurobehavioral, Biochemical, and Histological Approach in Male Wistar Rats. J. Mol. Neurosci. 2018, 66, 322–341. [Google Scholar] [CrossRef]
- Purushothaman, B.; Sumathi, T. 5,6,7-Trihydroxy Flavone Armoured Neurodegeneration Caused by Quinolinic Acid Induced Huntington’s Like Disease in Rat Striatum—Reinstating the Level of Brain Neurotrophins with Special Reference to Cognitive-Socio Behaviour, Biochemical and Histopathological Aspects. Neurosci. Res. 2022, 174, 25–35. [Google Scholar] [CrossRef]
- Pérez-De La Cruz, V.; González-Cortés, C.; Galván-Arzate, S.; Medina-Campos, O.N.; Pérez-Severiano, F.; Ali, S.F.; Pedraza-Chaverrí, J.; Santamaría, A. Excitotoxic Brain Damage Involves Early Peroxynitrite Formation in a Model of Huntington’s Disease in Rats: Protective Role of Iron Porphyrinate 5,10,15,20-Tetrakis (4-Sulfonatophenyl)Porphyrinate Iron (III). Neuroscience 2005, 135, 463–474. [Google Scholar] [CrossRef]
- Leipnitz, G.; Schumacher, C.; Scussiato, K.; Dalcin, K.B.; Wannmacher, C.M.; Wyse, A.T.; Dutra-Filho, C.S.; Wajner, M.; Latini, A. Quinolinic Acid Reduces the Antioxidant Defenses in Cerebral Cortex of Young Rats. Int. J. Dev. Neurosci. 2005, 23, 695–701. [Google Scholar] [CrossRef]
- Pérez-De La Cruz, V.; González-Cortés, C.; Pedraza-Chaverrí, J.; Maldonado, P.D.; Andrés-Martínez, L.; Santamaría, A. Protective Effect of S-Allylcysteine on 3-Nitropropionic Acid-Induced Lipid Peroxidation and Mitochondrial Dysfunction in Rat Brain Synaptosomes. Brain Res. Bull. 2006, 68, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Colle, D.; Hartwig, J.M.; Soares, F.A.; Farina, M. Probucol Modulates Oxidative Stress and Excitotoxicity in Huntington’s Disease Models in vitro. Brain Res. Bull. 2012, 87, 397–405. [Google Scholar] [CrossRef]
- Pérez-Severiano, F.; Ríos, C.; Segovia, J. Striatal Oxidative Damage Parallels the Expression of a Neurological Phenotype in Mice Transgenic for the Mutation of Huntington’s Disease. Brain Res. 2000, 862, 234–237. [Google Scholar] [CrossRef]
- Johri, A.; Calingasan, N.Y.; Hennessey, T.M.; Sharma, A.; Yang, L.; Wille, E.; Chandra, A.; Beal, M.F. Pharmacologic Activation of Mitochondrial Biogenesis Exerts Widespread Beneficial Effects in a Transgenic Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2012, 21, 1124–1137. [Google Scholar] [CrossRef]
- Chandra, A.; Sharma, A.; Calingasan, N.Y.; White, J.M.; Shurubor, Y.; Yang, X.W.; Beal, M.F.; Johri, A. Enhanced Mitochondrial Biogenesis Ameliorates Disease Phenotype in a Full-Length Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2016, 25, 2269–2282. [Google Scholar] [CrossRef]
- Brocardo, P.S.; McGinnis, E.; Christie, B.R.; Gil-Mohapel, J. Time-Course Analysis of Protein and Lipid Oxidation in the Brains of Yac128 Huntington’s Disease Transgenic Mice. Rejuvenation Res. 2016, 19, 140–148. [Google Scholar] [CrossRef]
- Askeland, G.; Rodinova, M.; Štufková, H.; Dosoudilova, Z.; Baxa, M.; Smatlikova, P.; Bohuslavova, B.; Klempir, J.; Nguyen, T.D.; Kuśnierczyk, A.; et al. A Transgenic Minipig Model of Huntington’s Disease Shows Early Signs of Behavioral and Molecular Pathologies. Dis. Model. Mech. 2018, 11, dmm035949. [Google Scholar]
- Dominah, G.A.; McMinimy, R.A.; Kallon, S.; Kwakye, G.F. Acute Exposure to Chlorpyrifos Caused NADPH Oxidase Mediated Oxidative Stress and Neurotoxicity in a Striatal Cell Model of Huntington’s Disease. Neurotoxicology 2017, 60, 54–69. [Google Scholar] [CrossRef]
- La Fontaine, M.A.; Geddes, J.W.; Banks, A.; Butterfield, D.A. 3-Nitropropionic Acid Induced in vivo Protein Oxidation in Striatal and Cortical Synaptosomes: Insights into Huntington’s Disease. Brain Res. 2000, 858, 356–362. [Google Scholar]
- Fontaine, M.A.; Geddes, J.W.; Banks, A.; Butterfield, D.A. Effect of Exogenous and Endogenous Antioxidants on 3-Nitropionic Acid-Induced in vivo Oxidative Stress and Striatal Lesions: Insights into Huntington’s Disease. J. Neurochem. 2000, 75, 1709–1715. [Google Scholar] [CrossRef]
- Souza, L.C.; Wilhelm, E.A.; Bortolatto, C.F.; Nogueira, C.W.; Boeira, S.P.; Jesse, C.R. Involvement of mGlu5 receptor in 3-nitropropionic acid-induced oxidative stress in rat striatum. Neurol. Res. 2014, 36, 833–840. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Orozco-Ibarra, M.; Chánez-Cárdenas, M.E.; Rangel-López, E.; Santamaría, A.; Pedraza-Chaverri, J.; Barrera-Oviedo, D.; Maldonado, P.D. Heme oxygenase-1 (HO-1) upregulation delays morphological and oxidative damage induced in an excitotoxic/pro-oxidant model in the rat striatum. Neuroscience 2013, 231, 91–101. [Google Scholar] [CrossRef]
- Antunes Wilhelm, E.; Ricardo Jesse, C.; Folharini Bortolatto, C.; Wayne Nogueira, C. Correlations between behavioural and oxidative parameters in a rat quinolinic acid model of Huntington’s disease: Protective effect of melatonin. Eur. J. Pharmacol. 2013, 701, 65–72. [Google Scholar] [CrossRef]
- Lou, S.; Lepak, V.C.; Eberly, L.E.; Roth, B.; Cui, W.; Zhu, X.H.; Öz, G.; Dubinsky, J.M. Oxygen consumption deficit in Huntington disease mouse brain under metabolic stress. Hum. Mol. Genet. 2016, 25, 2813–2826. [Google Scholar] [CrossRef]
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Torres, K.; Berríos, L.; Rosario, N.; Dufault, V.; Skatchkov, S.; Eaton, M.J.; Torres-Ramos, C.A.; Ayala-Torres, S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair 2009, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Chan, P.H. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J. Cereb. Blood Flow Metab. 2002, 22, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef]
- Chang, K.L.; New, L.S.; Mal, M.; Goh, C.W.; Aw, C.C.; Browne, E.R.; Chan, E.C. Metabolic profiling of 3-nitropropionic acid early-stage Huntington’s disease rat model using gas chromatography time-of-flight mass spectrometry. J. Proteome Res. 2011, 10, 2079–2087. [Google Scholar] [CrossRef]
- Pérez-Severiano, F.; Escalante, B.; Vergara, P.; Ríos, C.; Segovia, J. Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res. 2002, 951, 36–42. [Google Scholar] [CrossRef]
- Jang, M.; Cho, I.H. Sulforaphane Ameliorates 3-Nitropropionic Acid-Induced Striatal Toxicity by Activating the Keap1-Nrf2-ARE Pathway and Inhibiting the MAPKs and NF-κB Pathways. Mol. Neurobiol. 2016, 53, 2619–2635. [Google Scholar] [CrossRef]
- Aguilera, P.; Chánez-Cárdenas, M.E.; Floriano-Sánchez, E.; Barrera, D.; Santamaría, A.; Sánchez-González, D.J.; Pérez-Severiano, F.; Pedraza-Chaverrí, J.; Jiménez, P.D. Time-related changes in constitutive and inducible nitric oxide synthases in the rat striatum in a model of Huntington’s disease. Neurotoxicology 2007, 28, 1200–1207. [Google Scholar] [CrossRef]
- Napolitano, M.; Zei, D.; Centonze, D.; Palermo, R.; Bernardi, G.; Vacca, A.; Calabresi, P.; Gulino, A. NF-kB/NOS cross-talk induced by mitochondrial complex II inhibition: Implications for Huntington’s disease. Neurosci. Lett. 2008, 434, 241–246. [Google Scholar] [CrossRef]
- Petersen, M.H.; Willert, C.W.; Andersen, J.V.; Madsen, M.; Waagepetersen, H.S.; Skotte, N.H.; Nørremølle, A. Progressive Mitochondrial Dysfunction of Striatal Synapses in R6/2 Mouse Model of Huntington’s Disease. J. Huntingt. Dis. 2022, 11, 121–140. [Google Scholar] [CrossRef]
- Fernández, A.; Martínez-Ramírez, C.; Gómez, A.; de Diego, A.M.G.; Gandía, L.; Casarejos, M.J.; García, A.G. Mitochondrial dysfunction in chromaffin cells from the R6/1 mouse model of Huntington’s disease: Impact on exocytosis and calcium current regulation. Neurobiol. Dis. 2023, 179, 106046. [Google Scholar] [CrossRef]
- Lim, D.; Fedrizzi, L.; Tartari, M.; Zuccato, C.; Cattaneo, E.; Brini, M.; Carafoli, E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem. 2008, 283, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Rosenstock, T.R.; Cunha-Oliveira, T.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. Glutathione redox cycle dysregulation in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2012, 53, 1857–1867. [Google Scholar] [CrossRef]
- Frederick, N.M.; Bertho, J.; Patel, K.K.; Petr, G.T.; Bakradze, E.; Smith, S.B.; Rosenberg, P.A. Dysregulation of system xc(-) expression induced by mutant huntingtin in a striatal neuronal cell line and in R6/2 mice. Neurochem. Int. 2014, 76, 59–69. [Google Scholar] [CrossRef]
- Wang, J.Q.; Chen, Q.; Wang, X.; Wang, Q.C.; Wang, Y.; Cheng, H.P.; Guo, C.; Sun, Q.; Chen, Q.; Tang, T.S. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 2013, 288, 3070–3084. [Google Scholar] [CrossRef]
- Choi, Y.J.; Om, J.Y.; Kim, N.H.; Chang, J.E.; Park, J.H.; Kim, J.Y.; Lee, H.J.; Kim, S.S.; Chun, W. Heat Shock Transcription Factor-1 Suppresses Apoptotic Cell Death and ROS Generation in 3-Nitropropionic Acid-Stimulated Striatal Cells. Mol. Cell. Biochem. 2013, 375, 59–67. [Google Scholar] [CrossRef]
- Pruccoli, L.; Breda, C.; Teti, G.; Falconi, M.; Giorgini, F.; Tarozzi, A. Esculetin Provides Neuroprotection against Mutant Huntingtin-Induced Toxicity in Huntington’s Disease Models. Pharmaceuticals 2021, 14, 1044. [Google Scholar] [CrossRef]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of Copper Ion-Mediated Huntington’s Disease Progression. PLoS ONE 2007, 2, e334. [Google Scholar] [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLoS ONE 2013, 8, e77023. [Google Scholar] [CrossRef]
- Colle, D.; Santos, D.B.; Hartwig, J.M.; Godoi, M.; Braga, A.L.; Farina, M. Succinobucol versus Probucol: Higher Efficiency of Succinobucol in Mitigating 3-NP-Induced Brain Mitochondrial Dysfunction and Oxidative Stress in Vitro. Mitochondrion 2013, 13, 125–133. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; Abílio, V.C.; Frussa-Filho, R.; Kiyomoto, B.H.; Smaili, S.S. Old Mice Present Increased Levels of Succinate Dehydrogenase Activity and Lower Vulnerability to Dyskinetic Effects of 3-Nitropropionic Acid. Pharmacol. Biochem. Behav. 2009, 91, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, A.; Pérez-Severiano, F.; Rodríguez-Martínez, E.; Maldonado, P.D.; Pedraza-Chaverri, J.; Ríos, C.; Segovia, J. Comparative Analysis of Superoxide Dismutase Activity between Acute Pharmacological Models and a Transgenic Mouse Model of Huntington’s Disease. Neurochem. Res. 2001, 26, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, W.J.; Switonski, P.M.; Krzyzosiak, W.J.; Figlerowicz, M.; Figiel, M. Huntington Disease iPSCs Show Early Molecular Changes in Intracellular Signaling, the Expression of Oxidative Stress Proteins and the p53 Pathway. Dis. Model. Mech. 2015, 8, 1047–1057. [Google Scholar] [CrossRef]
- Maksimović, I.D.; Jovanović, M.D.; Colić, M.; Mihajlović, R.; Mićić, D.; Selaković, V.; Ninković, M.; Malicević, Z.; Rusić-Stojiljković, M.; Jovicić, A. Oxidative Damage and Metabolic Dysfunction in Experimental Huntington’s Disease: Selective Vulnerability of the Striatum and Hippocampus. Vojnosanit. Pregl. 2001, 58, 237–242. [Google Scholar]
- Tkác, I.; Keene, C.D.; Pfeuffer, J.; Low, W.C.; Gruetter, R. Metabolic Changes in Quinolinic Acid-Lesioned Rat Striatum Detected Non-Invasively by In Vivo ¹H NMR Spectroscopy. J. Neurosci. Res. 2001, 66, 891–898. [Google Scholar] [CrossRef]
- Choo, Y.S.; Mao, Z.; Johnson, G.V.; Lesort, M. Increased Glutathione Levels in Cortical and Striatal Mitochondria of the R6/2 Huntington’s Disease Mouse Model. Neurosci. Lett. 2005, 386, 63–68. [Google Scholar] [CrossRef]
- Hong, C.; Seo, H.; Kwak, M.; Jeon, J.; Jang, J.; Jeong, E.M.; Myeong, J.; Hwang, Y.J.; Ha, K.; Kang, M.J.; et al. Increased TRPC5 Glutathionylation Contributes to Striatal Neuron Loss in Huntington’s Disease. Brain 2015, 138, 3030–3047. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Ennis, M.D. Dysregulation of Ascorbate Release in the Striatum of Behaving Mice Expressing the Huntington’s Disease Gene. J. Neurosci. 2002, 22, RC202. [Google Scholar] [CrossRef]
- Shafie, A.; Ashour, A.A.; Anwar, S.; Anjum, F.; Hassan, M.I. Exploring molecular mechanisms, therapeutic strategies, and clinical manifestations of Huntington’s disease. Arch. Pharm. Res. 2024, 47, 571–595. [Google Scholar] [CrossRef]
- Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and Consistency. Mov. Disord. 1996, 11, 136–142. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Cárcamo-Fonfría, A.; Caballero-Muñoz, M.d.M.; Agúndez, J.A.G. Oxidative Stress in Huntington’s Disease. Biomolecules 2025, 15, 527. https://doi.org/10.3390/biom15040527
Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Cárcamo-Fonfría A, Caballero-Muñoz MdM, Agúndez JAG. Oxidative Stress in Huntington’s Disease. Biomolecules. 2025; 15(4):527. https://doi.org/10.3390/biom15040527
Chicago/Turabian StyleJiménez-Jiménez, Félix Javier, Hortensia Alonso-Navarro, Elena García-Martín, Alba Cárcamo-Fonfría, María del Mar Caballero-Muñoz, and José A. G. Agúndez. 2025. "Oxidative Stress in Huntington’s Disease" Biomolecules 15, no. 4: 527. https://doi.org/10.3390/biom15040527
APA StyleJiménez-Jiménez, F. J., Alonso-Navarro, H., García-Martín, E., Cárcamo-Fonfría, A., Caballero-Muñoz, M. d. M., & Agúndez, J. A. G. (2025). Oxidative Stress in Huntington’s Disease. Biomolecules, 15(4), 527. https://doi.org/10.3390/biom15040527