
A Pangenomic Approach to Improve Population Genetics Analysis and Reference Bias in Underrepresented Middle Eastern and Horn of Africa Populations

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Building the Pangenome Graph
2.2. Mapping and Variant Calling
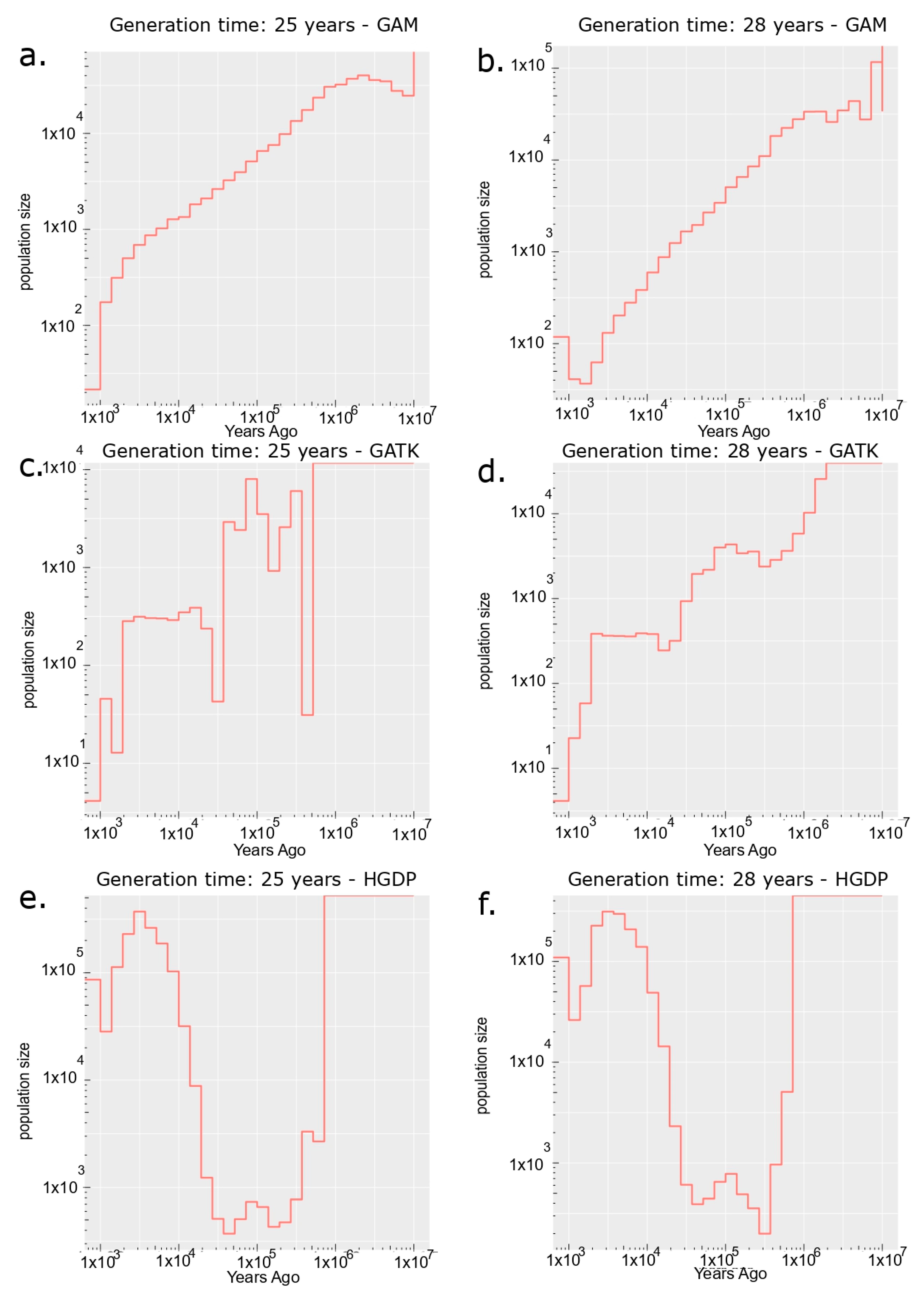
2.3. Effective Population Size Estimates and Simulations
2.4. Metrics of Variant Call Assessment
3. Results
3.1. Ancestry Analysis for Variation Graph Construction
3.2. Analysis of Variant Caller
3.3. Effective Population Size
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- López, S.; van Dorp, L.; Hellenthal, G. Human Dispersal Out of Africa: A Lasting Debate. Evol. Bioinform. Online 2016, 11, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Pagani, L.; Schiffels, S.; Gurdasani, D.; Danecek, P.; Scally, A.; Chen, Y.; Xue, Y.; Haber, M.; Ekong, R.; Oljira, T.; et al. Tracing the Route of Modern Humans out of Africa by Using 225 Human Genome Sequences from Ethiopians and Egyptians. Am. J. Hum. Genet. 2015, 96, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Henn, B.M.; Botigué, L.R.; Gravel, S.; Wang, W.; Brisbin, A.; Byrnes, J.K.; Fadhlaoui-Zid, K.; Zalloua, P.A.; Moreno-Estrada, A.; Bertranpetit, J.; et al. Genomic Ancestry of North Africans Supports Back-to-Africa Migrations. PLoS Genet. 2012, 8, e1002397. [Google Scholar] [CrossRef]
- Molinaro, L.; Montinaro, F.; Yelmen, B.; Marnetto, D.; Behar, D.M.; Kivisild, T.; Pagani, L. West Asian Sources of the Eurasian Component in Ethiopians: A Reassessment. Sci. Rep. 2019, 9, 18811. [Google Scholar] [CrossRef] [PubMed]
- Almarri, M.A.; Haber, M.; Lootah, R.A.; Hallast, P.; Al Turki, S.; Martin, H.C.; Xue, Y.; Tyler-Smith, C. The Genomic History of the Middle East. Cell 2021, 184, 4612. [Google Scholar] [CrossRef]
- Rodriguez-Flores, J.L.; Fakhro, K.; Agosto-Perez, F.; Ramstetter, M.D.; Arbiza, L.; Vincent, T.L.; Robay, A.; Malek, J.A.; Suhre, K.; Chouchane, L.; et al. Indigenous Arabs Are Descendants of the Earliest Split from Ancient Eurasian Populations. Genome Res. 2016, 26, 151–162. [Google Scholar] [CrossRef]
- Tobler, R.; Souilmi, Y.; Huber, C.D.; Bean, N.; Turney, C.S.M.; Grey, S.T.; Cooper, A. The Role of Genetic Selection and Climatic Factors in the Dispersal of Anatomically Modern Humans out of Africa. Proc. Natl. Acad. Sci. USA 2023, 120, e2213061120. [Google Scholar] [CrossRef]
- Alsmadi, O.; Thareja, G.; Alkayal, F.; Rajagopalan, R.; John, S.E.; Hebbar, P.; Behbehani, K.; Thanaraj, T.A. Genetic Substructure of Kuwaiti Population Reveals Migration History. PLoS ONE 2013, 8, e74913. [Google Scholar] [CrossRef]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic Insights into the Origin of Farming in the Ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef]
- Martiniano, R.; Haber, M.; Almarri, M.A.; Mattiangeli, V.; Kuijpers, M.C.M.; Chamel, B.; Breslin, E.M.; Littleton, J.; Almahari, S.; Aloraifi, F.; et al. Ancient Genomes Illuminate Eastern Arabian Population History and Adaptation against Malaria. Cell Genom. 2024, 4, 100507. [Google Scholar] [CrossRef]
- Haber, M.; Gauguier, D.; Youhanna, S.; Patterson, N.; Moorjani, P.; Botigué, L.R.; Platt, D.E.; Matisoo-Smith, E.; Soria-Hernanz, D.F.; Spencer Wells, R.; et al. Genome-Wide Diversity in the Levant Reveals Recent Structuring by Culture. PLoS Genet. 2013, 9, e1003316. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.A.; Mulligan, C.J.; Al-Meeri, A.; Raaum, R.L. Early Back-to-Africa Migration into the Horn of Africa. PLoS Genet. 2014, 10, e1004393. [Google Scholar] [CrossRef]
- Fregel, R.; Méndez, F.L.; Bokbot, Y.; Martín-Socas, D.; Camalich-Massieu, M.D.; Santana, J.; Morales, J.; Ávila-Arcos, M.C.; Underhill, P.A.; Shapiro, B.; et al. Ancient Genomes from North Africa Evidence Prehistoric Migrations to the Maghreb from Both the Levant and Europe. Proc. Natl. Acad. Sci. USA 2018, 115, 6774–6779. [Google Scholar] [CrossRef] [PubMed]
- Serradell, J.M.; Lorenzo-Salazar, J.M.; Flores, C.; Lao, O.; Comas, D. Modelling the Demographic History of Human North African Genomes Points to a Recent Soft Split Divergence between Populations. Genome Biol. 2024, 25, 201. [Google Scholar] [CrossRef]
- Ambrose, S.H. Chronology of the Later Stone Age and Food Production in East Africa. J. Archaeol. Sci. 1998, 25, 377–392. [Google Scholar] [CrossRef]
- Fortes-Lima, C.A.; Burgarella, C.; Hammarén, R.; Eriksson, A.; Vicente, M.; Jolly, C.; Semo, A.; Gunnink, H.; Pacchiarotti, S.; Mundeke, L.; et al. The Genetic Legacy of the Expansion of Bantu-Speaking Peoples in Africa. Nature 2023, 625, 540–547. [Google Scholar] [CrossRef]
- Shriner, D.; Tekola-Ayele, F.; Adeyemo, A.; Rotimi, C.N. Ancient Human Migration after Out-of-Africa. Sci. Rep. 2016, 6, 26565. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Sánchez, M.; Abdeli, A.; Bekada, A.; Calafell, F.; Benhassine, T.; Comas, D. The Impact of Recent Demography on Functional Genetic Variation in North African Human Groups. Mol. Biol. Evol. 2023, 41, msad283. [Google Scholar] [CrossRef] [PubMed]
- Vilà-Valls, L.; Abdeli, A.; Lucas-Sánchez, M.; Bekada, A.; Calafell, F.; Benhassine, T.; Comas, D. Understanding the Genomic Heterogeneity of North African Imazighen: From Broad to Microgeographical Perspectives. Sci. Rep. 2024, 14, 9979. [Google Scholar] [CrossRef]
- Ali, A.A.; Aalto, M.; Jonasson, J.; Osman, A. Genome-Wide Analyses Disclose the Distinctive HLA Architecture and the Pharmacogenetic Landscape of the Somali Population. Sci. Rep. 2020, 10, 5652. [Google Scholar] [CrossRef]
- Al-Sabti, H.; Al-Hinai, A.T.; Al-Zakwani, I.; Al-Hashmi, K.; Al Mahmeed, W.; Arafah, M.; Shehab, A.; Al Tamimi, O.; Al Awadhi, M.; Al Herz, S.; et al. The Achievement of Non-High-Density Lipoprotein Cholesterol Target in Patients with Very High Atherosclerotic Cardiovascular Disease Risk Stratified by Triglyceride Levels Despite Statin-Controlled Low-Density Lipoprotein Cholesterol. Oman Med. J. 2022, 37, e367. [Google Scholar] [CrossRef]
- Westgard, B.; Martinson, B.C.; Maciosek, M.; Brown, M.; Xu, Z.; Farah, F.; Ahmed, O.; Dalmar, A.; Dubois, D.; Sanka, L.; et al. Prevalence of Cardiovascular Disease and Risk Factors Among Somali Immigrants and Refugees. J. Immigr. Minor. Health 2021, 23, 680–688. [Google Scholar] [CrossRef]
- Berg, J.J.; Harpak, A.; Sinnott-Armstrong, N.; Joergensen, A.M.; Mostafavi, H.; Field, Y.; Boyle, E.A.; Zhang, X.; Racimo, F.; Pritchard, J.K.; et al. Reduced Signal for Polygenic Adaptation of Height in UK Biobank. eLife 2019, 8, e39725. [Google Scholar] [CrossRef]
- Sohail, M.; Maier, R.M.; Ganna, A.; Bloemendal, A.; Martin, A.R.; Turchin, M.C.; Chiang, C.W.; Hirschhorn, J.; Daly, M.J.; Patterson, N.; et al. Polygenic Adaptation on Height Is Overestimated due to Uncorrected Stratification in Genome-Wide Association Studies. eLife 2019, 8, e39702. [Google Scholar] [CrossRef]
- Hingorani, A.D.; Gratton, J.; Finan, C.; Schmidt, A.F.; Patel, R.; Sofat, R.; Kuan, V.; Langenberg, C.; Hemingway, H.; Morris, J.K.; et al. Performance of Polygenic Risk Scores in Screening, Prediction, and Risk Stratification: Secondary Analysis of Data in the Polygenic Score Catalog. BMJ Med. 2023, 2, e000554. [Google Scholar] [CrossRef]
- Sherman, R.M.; Forman, J.; Antonescu, V.; Puiu, D.; Daya, M.; Rafaels, N.; Boorgula, M.P.; Chavan, S.; Vergara, C.; Ortega, V.E.; et al. Assembly of a Pan-Genome from Deep Sequencing of 910 Humans of African Descent. Nat. Genet. 2018, 51, 30–35. [Google Scholar] [CrossRef]
- Tetikol, H.S.; Turgut, D.; Narci, K.; Budak, G.; Kalay, O.; Arslan, E.; Demirkaya-Budak, S.; Dolgoborodov, A.; Kabakci-Zorlu, D.; Semenyuk, V.; et al. Pan-African Genome Demonstrates How Population-Specific Genome Graphs Improve High-Throughput Sequencing Data Analysis. Nat. Commun. 2022, 13, 4384. [Google Scholar] [CrossRef]
- Garrison, E.; Sirén, J.; Novak, A.M.; Hickey, G.; Eizenga, J.M.; Dawson, E.T.; Jones, W.; Garg, S.; Markello, C.; Lin, M.F.; et al. Variation Graph Toolkit Improves Read Mapping by Representing Genetic Variation in the Reference. Nat. Biotechnol. 2018, 36, 875–879. [Google Scholar] [CrossRef]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into Human Genetic Variation and Population History from 929 Diverse Genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef]
- Mallick, S.; Micco, A.; Mah, M.; Ringbauer, H.; Lazaridis, I.; Olalde, I.; Patterson, N.; Reich, D. The Allen Ancient DNA Resource (AADR): A Curated Compendium of Ancient Human Genomes. Sci. Data 2024, 11, 182. [Google Scholar] [CrossRef]
- Harney, É.; Patterson, N.; Reich, D.; Wakeley, J. Assessing the Performance of qpAdm: A Statistical Tool for Studying Population Admixture. Genetics 2021, 217, iyaa045. [Google Scholar] [CrossRef]
- Perez, G.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser Database: 2025 Update. Nucleic Acids Res. 2025, 53, D1243–D1249. [Google Scholar] [CrossRef]
- Martiniano, R.; Garrison, E.; Jones, E.R.; Manica, A.; Durbin, R. Removing Reference Bias and Improving Indel Calling in Ancient DNA Data Analysis by Mapping to a Sequence Variation Graph. Genome Biol. 2020, 21, 250. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Speidel, L.; Forest, M.; Shi, S.; Myers, S.R. A Method for Genome-Wide Genealogy Estimation for Thousands of Samples. Nat. Genet. 2019, 51, 1321–1329. [Google Scholar] [CrossRef]
- Fernandes, V.; Triska, P.; Pereira, J.B.; Alshamali, F.; Rito, T.; Machado, A.; Fajkošová, Z.; Cavadas, B.; Černý, V.; Soares, P.; et al. Genetic Stratigraphy of Key Demographic Events in Arabia. PLoS ONE 2015, 10, e0118625. [Google Scholar] [CrossRef]
- Excoffier, L.; Foll, M. Fastsimcoal: A Continuous-Time Coalescent Simulator of Genomic Diversity under Arbitrarily Complex Evolutionary Scenarios. Bioinformatics 2011, 27, 1332–1334. [Google Scholar] [CrossRef]
- Excoffier, L.; Marchi, N.; Marques, D.A.; Matthey-Doret, R.; Gouy, A.; Sousa, V.C. fastsimcoal2: Demographic Inference under Complex Evolutionary Scenarios. Bioinformatics 2021, 37, 4882–4885. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Hebbar, P.; Abu-Farha, M.; Alkayal, F.; Nizam, R.; Elkum, N.; Melhem, M.; John, S.E.; Channanath, A.; Abubaker, J.; Bennakhi, A.; et al. Genome-Wide Association Study Identifies Novel Risk Variants from RPS6KA1, CADPS, VARS, and DHX58 for Fasting Plasma Glucose in Arab Population. Sci. Rep. 2020, 10, 152. [Google Scholar] [CrossRef]
- Maier, R.; Flegontov, P.; Flegontova, O.; Işıldak, U.; Changmai, P.; Reich, D. On the Limits of Fitting Complex Models of Population History to F-Statistics. eLife 2023, 12, e85492. [Google Scholar] [CrossRef]
- Koenig, Z.; Yohannes, M.T.; Nkambule, L.L.; Zhao, X.; Goodrich, J.K.; Kim, H.A.; Wilson, M.W.; Tiao, G.; Hao, S.P.; Sahakian, N.; et al. A Harmonized Public Resource of Deeply Sequenced Diverse Human Genomes. Genome Res. 2024, 34, 796–809. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L. The Human Genome Diversity Project: Past, Present and Future. Nat. Rev. Genet. 2005, 6, 333–340. [Google Scholar] [CrossRef]
- Hickey, G.; Heller, D.; Monlong, J.; Sibbesen, J.A.; Sirén, J.; Eizenga, J.; Dawson, E.T.; Garrison, E.; Novak, A.M.; Paten, B. Genotyping Structural Variants in Pangenome Graphs Using the vg Toolkit. Genome Biol. 2020, 21, 35. [Google Scholar] [CrossRef]
- Liao, W.-W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A Draft Human Pangenome Reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
- Nassir, N.; Almarri, M.A.; Kumail, M.; Mohamed, N.; Balan, B.; Hanif, S.; AlObathani, M.; Jamalalail, B.; Elsokary, H.; Kondaramage, D.; et al. A Draft Arab Pangenome Reference. bioRxiv 2024. [Google Scholar] [CrossRef]
- Omberg, L.; Salit, J.; Hackett, N.; Fuller, J.; Matthew, R.; Chouchane, L.; Rodriguez-Flores, J.L.; Bustamante, C.; Crystal, R.G.; Mezey, J.G. Inferring Genome-Wide Patterns of Admixture in Qataris Using Fifty-Five Ancestral Populations. BMC Genet. 2012, 13, 49. [Google Scholar] [CrossRef]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and Estimation of Effective Population Size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef]
- Stadler, T.; Vaughan, T.G.; Gavryushkin, A.; Guindon, S.; Kühnert, D.; Leventhal, G.E.; Drummond, A.J. How Well Can the Exponential-Growth Coalescent Approximate Constant-Rate Birth-Death Population Dynamics? Proc. Biol. Sci. 2015, 282, 20150420. [Google Scholar] [CrossRef]
- Speidel, L.; Cassidy, L.; Davies, R.W.; Hellenthal, G.; Skoglund, P.; Myers, S.R. Inferring Population Histories for Ancient Genomes Using Genome-Wide Genealogies. Mol. Biol. Evol. 2021, 38, 3497–3511. [Google Scholar] [CrossRef]
- Markus, B.; Alshafee, I.; Birk, O.S. Deciphering the Fine-Structure of Tribal Admixture in the Bedouin Population Using Genomic Data. Heredity 2014, 112, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, T.; Xue, Y.; Evison, M.; Tyler-Smith, C. Genetic Structure of Nomadic Bedouin from Kuwait. Heredity 2009, 103, 425. [Google Scholar] [CrossRef] [PubMed]
- Na’amnih, W.; Romano-Zelekha, O.; Kabaha, A.; Rubin, L.P.; Bilenko, N.; Jaber, L.; Honovich, M.; Shohat, T. Prevalence of Consanguineous Marriages and Associated Factors among Israeli Bedouins. J. Community Genet. 2014, 5, 395–398. [Google Scholar] [CrossRef]
- Singer, S.; Davidovitch, N.; Abu Fraiha, Y.; Abu Freha, N. Consanguinity and Genetic Diseases among the Bedouin Population in the Negev. J. Community Genet. 2020, 11, 13–19. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.; Yun, T.; Moreno, M.; Li, H.; Litterman, N.; Kolesnikov, A.; Noblin, E.; Chang, P.-C.; Shastri, A.; Dorfman, E.H.; et al. A Population-Specific Reference Panel for Improved Genotype Imputation in African Americans. Commun. Biol. 2021, 4, 1269. [Google Scholar] [CrossRef]
- Sengupta, D.; Botha, G.; Meintjes, A.; Mbiyavanga, M.; AWI-Gen Study; H3Africa Consortium; Hazelhurst, S.; Mulder, N.; Ramsay, M.; Choudhury, A. Performance and Accuracy Evaluation of Reference Panels for Genotype Imputation in Sub-Saharan African Populations. Cell Genom. 2023, 3, 100332. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L.; Daviglus, M.L.; Durazo-Arvizu, R.A.; Schneiderman, N.; Kaplan, R.C.; Laurie, C.C. Ancestry-Specific Recent Effective Population Size in the Americas. PLoS Genet. 2018, 14, e1007385. [Google Scholar] [CrossRef]
- Thareja, G.; Al-Sarraj, Y.; Belkadi, A.; Almotawa, M.; Qatar Genome Program Research (QGPR) Consortium; Suhre, K.; Albagha, O.M.E. Whole Genome Sequencing in the Middle Eastern Qatari Population Identifies Genetic Associations with 45 Clinically Relevant Traits. Nat. Commun. 2021, 12, 1250. [Google Scholar] [CrossRef]
- Elashi, A.A.; Toor, S.M.; Umlai, U.-K.I.; Al-Sarraj, Y.A.; Taheri, S.; Suhre, K.; Abou-Samra, A.B.; Albagha, O.M.E. Genome-Wide Association Study and Trans-Ethnic Meta-Analysis Identify Novel Susceptibility Loci for Type 2 Diabetes Mellitus. BMC Med. Genom. 2024, 17, 115. [Google Scholar]
- Novembre, J.; Stephens, M. Interpreting Principal Component Analyses of Spatial Population Genetic Variation. Nat. Genet. 2008, 40. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Target | Source Populations | p-Value |
|---|---|---|
| Somali | Kenya Kansyore 3300 BP (53.0%), Mozabite (47.0%) | 0.524 |
| Somali | Kenya_IA_Pastoral (27.0%), Ethiopian_BetaIsrael (Ethiopian Jews) (73.0%) | 0.189 |
| Somali | LWK (86.7%), Mozabite (5.2%) Kenya IA (8.1%) | 3.08 × 10−46 |
| Mozabite | MSL (31.8%), Palestinian (68.2%) | 0.347 |
| Mozabite | MSL (19.0%), Bedouin (81.0%) | 0.453 |
| Bedouin | Palestinian (69.0%), Mozabite (31.0%) | 0.377 |
| Bedouin | Palestinian (62.0%), Mozabite (38.0%) | 0.389 |
| Dataset | Generation Time | Ne Estimation: Empirical Data (Relate) | Ne Simulated Estimation: FastSimCoal v28 (95% CI) |
|---|---|---|---|
| GAM | 25 | 16 | 17 (95% CI: 15–19) |
| GAM | 28 | 125 | 34 (95% CI: 28–35) |
| HGDP | 25 | 79,168 | 3295 (95% CI: 324–3468) |
| HGDP | 28 | 110,485 | 9639 (95% CI: 5164–78,179) |
| GATK | 25 | 4 | 13 (95% CI: 12–14) |
| GATK | 28 | 3 | 14 (95% CI: 11–15) |
| Variant Allele Frequency Comparisons | p-Value (Direction of Frequency Distribution Shift) |
|---|---|
| GAM vs. GATK | 1.76 × 10−3 (GAM > GATK) |
| GAM vs. HGDP | <2.2 × 10−16 (GAM > HGDP) |
| GATK vs. HGDP | <2.2 × 10−16 (GATK > HGDP) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, A.; Foare, R.; Campbell, P.; Twine, N.A.; Bauer, D.C.; Johar, A.S. A Pangenomic Approach to Improve Population Genetics Analysis and Reference Bias in Underrepresented Middle Eastern and Horn of Africa Populations. Biomolecules 2025, 15, 582. https://doi.org/10.3390/biom15040582
Oliva A, Foare R, Campbell P, Twine NA, Bauer DC, Johar AS. A Pangenomic Approach to Improve Population Genetics Analysis and Reference Bias in Underrepresented Middle Eastern and Horn of Africa Populations. Biomolecules. 2025; 15(4):582. https://doi.org/10.3390/biom15040582
Chicago/Turabian StyleOliva, Adrien, Rachel Foare, Peter Campbell, Natalie A. Twine, Denis C. Bauer, and Angad Singh Johar. 2025. "A Pangenomic Approach to Improve Population Genetics Analysis and Reference Bias in Underrepresented Middle Eastern and Horn of Africa Populations" Biomolecules 15, no. 4: 582. https://doi.org/10.3390/biom15040582
APA StyleOliva, A., Foare, R., Campbell, P., Twine, N. A., Bauer, D. C., & Johar, A. S. (2025). A Pangenomic Approach to Improve Population Genetics Analysis and Reference Bias in Underrepresented Middle Eastern and Horn of Africa Populations. Biomolecules, 15(4), 582. https://doi.org/10.3390/biom15040582